|
|
 |
|
|
|
|
|
Light and Electron Microscopy of Macular Corneal Dystrophy: A Case Study
Digital Journal of Ophthalmology 2004
Volume 10, Number 1
March 18, 2004
|
Printer Friendly
|
 Daniel Gore, M.D.
Daniel Gore, M.D. | Mount Sinai School of Medicine Shamim Ahmed Haji, M.D. | Mount Sinai School of Medicine Aarthi Balashanmugam | Mount Sinai School of Medicine Ivo Dualan, M.D. | Mount Sinai School of Medicine Ronald Gordon, Ph.D | Mount Sinai School of Medicine Alan Friedman, M.D. | Mount Sinai School of Medicine Penny Asbell, M.D. | Mount Sinai School of Medicine
|
|
|
| Abstract | Objective
To report a case of macular corneal dystrophy requiring bilateral corneal transplantation.
Methods
A 24 year-old female presented with progressive loss of vision over a period of 14 years. Originally described as ‘fogginess’, the patient complained of appreciable deterioration in the past six months, with one episode of redness, tearing, and photophobia bilaterally. Visual acuity was recorded as 20/80 OU. Slit lamp examination revealed diffusely hazy corneas with opacities bilaterally. Bilateral irregular astigmatism was evidenced topographically. The patient subsequently underwent consecutive penetrating keratoplasty. Light microscopy and transmission electron microscopy were performed on the removed corneal specimens.
Results
Light microscopy of the corneas revealed deposits of glycosaminoglycans in keratocytes, endothelial cells and extracellularly within the stroma, all staining positively with Alcian blue and periodic acid-Schiff. Electron microscopy showed keratocytes distended by membrane bound intracytoplasmic vacuoles containing electron dense fibrillogranular material. Some of the vacuoles contained dense osmiophilic whirls. Similar membrane bound, intracytoplasmic vacuoles containing fibrillogranular material were present in the endothelium keratocytes, in endothelial cells, and in small pools lying extracellularly within and between stromal lamellae, which stained with Alcian blue and periodic acid.
Conclusion
This report highlights one of the more aggressive forms of corneal dystrophy, manifesting in the first decade and characteristically impoverishing vision by the second. In addition, we review the principle histological features associated with this disorder.
Keywords
cornea, dystrophy, keratoplasty, glycosaminoglycan | | | Introduction | INTRODUCTION
Macular corneal dystrophy (MCD) is an autosomal-recessive disorder in which patients in the first decade of life develop fine, diffuse, superficial corneal clouding in the central stroma. By their teens, these opacities extend to the periphery involving the full thickness of the cornea, characteristically more superficial and prominent in the center, and deeper and more discrete in the periphery. Slowly, progressive loss of functional vision ensues, usually requiring surgical restoration by penetrating keratoplasty.
We report light and electron microscopy findings in a patient with a chronic history of deteriorating vision who underwent bilateral corneal graft surgery.
CASE REPORT
A 24 year-old female presented to our clinic with a 14-year history of bilateral corneal opacity. She complained of ‘foggy’ vision, particularly in the morning, which had noticeably worsened in the past six months. Two months prior, she had experienced a five-day episode of redness, tearing, and photophobia in both eyes.
Her previous ocular history began at age 10 when she complained of trouble reading clearly. She had no medical or surgical history of note. On examination done elsewhere, uncorrected visual acuities were recorded as 20/30 OD and 20/25 OS. On slit lamp examination the epithelium appeared clear though slight irregularity was noted on fluorescein staining. Multiple, predominantly anterior, stromal "crystalline" opacities were noted. The anterior chamber and lens were clear OU. The impression at the time was of a crystalline dystrophy. At follow-up a year later, visual acuities were unchanged, tonometry was within limits, and fundus exam was normal.
At age 20, the patient complained of photophobia, with a visual acuity of 20/40 OD and 20/30 OS noted elsewhere. Corneal examination revealed a well demarcated superficial stromal opacity with adjacent corneal surface exhibiting a lack of smoothness and discrete opacities. The corneal endothelium was noted as normal. A diagnosis of macular corneal dystrophy was made. Despite symptoms, the patient was able to continue working as an accountant at a global bank.
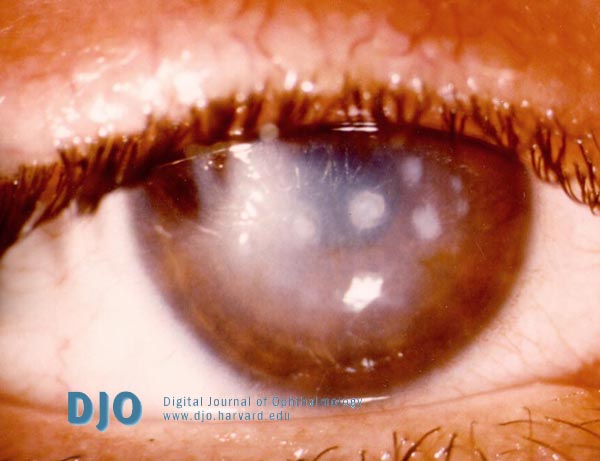
At age 24, visual acuity had deteriorated to 20/80 OU. Slit lamp examination revealed diffusely hazy corneas with opacities bilaterally (Figure 1). Corneal thickness was measured to be 568µm OD and 392µm OS, though the accuracy of these values are questionable since the patient had difficulty cooperating with these measurements due to photophobia and anxiety. Topography revealed bilateral irregular astigmatism.
After experiencing increased fogginess of vision and photophobia, the patient decided to undergo consecutive penetrating keratoplasty at age 25. The post-operative period was unremarkable for both eyes. The most recent post-surgical manifest refraction was 20/25 OD (one year post-op.) and 20/40 OS (five months post-op.) with clear corneas throughout OU. The patient subjectively reports excellent vision and has changed careers and now works in theater.
| |

Figure 1
Slit lamp view of the right cornea with macular dystrophy demonstrating central haziness of the stroma with multiple irregular grey-white opacities.
|
|
| Materials and Methods | Hemotoxylin and eosin, Alcian blue, periodic acid-Schiff (PAS), trichrome, and congo red stains were performed on the specimen using standard techniques.
For transmission electron microscopy, the corneal specimen was immediately immersed in a fixative solution containing 3% glutaraldehyde with 0.2 M sodium cacodylate at pH 7.4. After overnight fixation, the fixative solution was removed and replaced with a phosphate buffer followed by 1% osmium tetroxide buffered with sodium cacodylate. After one hour, the osmium was replaced with increasing concentrations of ethanol through propylene oxide and into embed 812. The tissue was oriented in the epon block to cut longitudinal sections. One micrometer plastic sections were cut, stained with methyl blue and azure II and observed with light microscopy. Representative areas were chosen for ultrathin sectioning and observed with a JEM 100CX transmission electron microscope (JEOL, LTD. Tokyo, Japan).
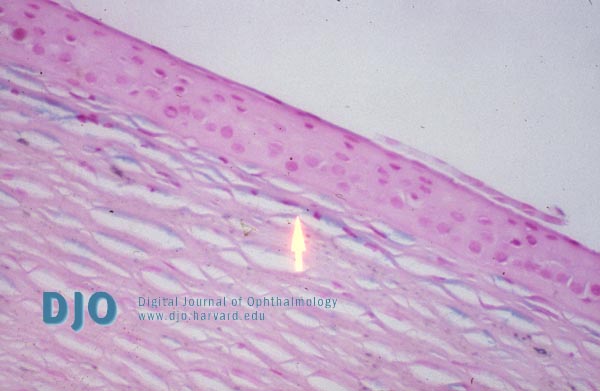
| | | Results | Light microscopy of the excised left cornea showed a normal epithelium and Bowman's membrane. Hemotoxylin and eosin staining revealed faintly basophilic deposits. These deposits stained positively for glycosaminoglycans with Alcian blue in keratocytes, in endothelial cells, and in small pools lying extracellularly within and between stromal lamellae (Figure 2). These deposits are PAS positive, trichrome negative, and congo red negative. Descemet's membrane was normal. They were not birefringent in polarized light.
Within the corneal stroma, electron microscopy revealed keratocytes distended by membrane bound intracytoplasmic vacuoles containing electron dense fibrillogranular material (Figure 3). A few of the vacuoles contained dense osmiophilic whirls. Similar membrane bound, intracytoplasmic vacuoles containing fibrillogranular material were present in the endothelium.
| |

Figure 2
Light microscopic appearance of corneal macular dystrophy showing Alcian Blue-stained granular deposits (arrow) irregularly throughout the entire stroma.
|
|

Figure 3
Transmission electron microscopy (magnification 3000X; inset 20,000x) of corneal macular dystrophy illustrating keratocytes distended by membrane bound intracytoplasmic vacuoles (1A, 1B - low and high power views) containing electron dense fibrillogranular material. 2 - membranous extracellular osmiophilic whorl.
|
|
| Discussion | This report highlights one of the more aggressive forms of corneal dystrophy, manifesting in the first decade and characteristically impoverishing vision by the second. We describe a case of macular corneal dystrophy with slit lamp examination and pathological findings similar to those found in the literature. Slit lamp examination in this case revealed diffusely hazy corneas with small, irregular, grey-white opacities bilaterally. Light microscopy indicated deposits of abnormal glycosaminoglycans in keratocytes, endothelial cells, and in small pools lying extracellularly within and between stromal lamellae, which stained positively with Alcian blue and PAS. Electron microscopy revealed keratocytes distended by membrane bound intracytoplasmic vacuoles containing electron dense fibrillogranular material. Some of the vacuoles contained dense osmiophilic whirls. Similar membrane bound, intracytoplasmic vacuoles containing fibrillogranular material were present in the endothelium.
Macular corneal dystrophy (MCD), an autosomal-recessive disease, represents the least common of the established corneal stromal dystrophies. In keeping with classical Mendelian inheritance heterozygous carriers are not phenotypic, and parents of homozygotes, if not overtly consanguineous, can often be shown to be of common ancestry [1].
MCD initially presents in the first decade of life with gradual bilateral loss of vision. Initial changes include a fine stromal opacity extending centrifugally to the limbus which, by the second decade, has usually encompassed the full thickness of the cornea including the endothelium. With time the opacity increases in density and is accompanied by dense superficial macules, which elevate the corneal surface and contribute to visual loss [2]. Painful recurrent erosions, frequently seen in lattice dystrophy, are a less common, though by no means less debilitating feature of this disease. Photophobia is also recognized and often appears out of proportion with corneal involvement. Significant failure of vision by the second to third decade is an inevitable consequence, indicating keratoplasty [2].
Slit lamp examination early on in the disease reveals a central ground-glass haze in the superficial stroma. Within this haziness are denser grey-white macular opacities with indistinct borders which can protrude anteriorly distorting overlying epithelium, or posteriorly producing a guttate appearance at Descemet’s membrane [3]. With time these opacities may enlarge and coalesce increasing surface irregularity. However, in contrast to corneal thickening associated with other corneal dystrophies [4], pachymetry reveals significant central thinning, resulting in astigmatism.
Light microscopy of MCD shows abnormal deposits of glycosaminoglycans in Bowman’s histiocytes, keratocytes, between the stromal lamellae, Descemet’s membrane, and endothelium. These glycosaminoglycans stain positively with Alcian blue and periodic acid-Schiff [5,6].
Electron microscopic findings, first described by Klintworth and Vogel (1964) [7], reveal these deposits correspond to electro-lucent fibrillogranular material visible within membrane-bound intracytoplasmic vacuoles. Within the distended layers observed on light microscopy, numerous such vacuoles swell keratocytes, often coalescing to impart an engorged appearance and honeycombed structure to the cytoplasm. In the stroma, Descemet's membrane and endothelium, similar material is additionally seen without a limiting membrane as osmiophilic whorls. The posterior layer of Descemet's membrane is sometimes seen to contain numerous focal thickenings that resemble corneal guttata. Dilated cisternae of rough-endoplasmic reticulum containing a granular material are found in both stroma and endothelium, although uncertainty exists as to whether they can be positively identified as the origin of the intracellular vacuoles [5].
Histologically, such abnormalities have been shown to be the sequelae of an error in metabolism of glycosaminoglycans within the cornea, in particular the proteoglycan keratan sulfate, resulting in its abnormal intra-and extracellular deposition [8]. These collections, staining with, amongst others, Alcian blue, form the basis of MCD classification, two major forms of which exist. This is based on measurements of the serum level of sulfated keratan sulfate (KS), the dominant glycosaminoglycan expressed in the adult cornea, as determined by antibodies directed against sulfated KS using enzyme-linked immunosorbent assay (ELISA) [9]. In Type I, accounting for the majority of patients, neither the cornea nor the serum contains normally sulfated KS, whereas in Type II there are low or normal amounts of antigenic KS in both the serum and the cornea 10. A subtype, IA, as yet distinct to Saudi Arabia and Germany, describes absent sulfated KS in the cornea and serum, but which is present in the keratocytes [10,11].
Recently, researchers have identified a new carbohydrate sulphotransferase gene (CHST6), located on chromosome 16 (16q22), encoding an enzyme designated corneal N-acetylglucosamine-6-sulphotransferase (C-GlcNac6ST) [12]. This gene product is thought to be important in the production of sulphated KS. Lumican, a KS proteoglycan present in large quantities in the corneal stroma interacts with collagen fibrils and helps maintain their size and ordered structure as well as corneal transparency [13]. The production of unsulfated KS chains attached to Lumican in the corneas of type I MCD, as a consequence of a lack of CHST6 activity, causes a loss of such interstitial structure, thereby reducing transparency in the corneas of MCD patients [14]. An analogous mechanism for Type II MCD, with normal amounts of sulphated KS in both the cornea (and serum) remains unclear.
Despite the use of tinted lenses to reduce photophobia and appropriate medical management of sporadic erosions, corneal replacement will almost inevitably be warranted to return function back to the patient. However, as with other corneal dystrophies, recurrence can occur. One study (41 transplants, 31 eyes, 16 patients) showed six eyes of four patients had repeat keratoplasty because of clinical recurrence and visual impairment, confirmed pathologically in all but one [15].
Future studies, including genetic, of macular corneal dystrophy may lead to improved diagnostic and therapeutic options.
| | | References | 1. Jonasson, F., Johannsson ,JH., Garner, A., Rice, NS., Macular Corneal Dystrophy in Iceland. Eye 3(pt 4):446-54 (1989).
2. Bron, AJ., Genetics of the Corneal Dystrophies: What We Have Learned in the Past Twenty-five Years. Cornea19(5):699–711 (2000).
3. Arffa, RC. ed., Grayson’s Diseases of the Cornea. St. Louis: Mosby (1997).
4. Donnenfeld, ED., Cohen, EJ., Ingraham, HJ., Poleski, SA., Goldsmith, E., Laibson, PR., Corneal Thinning in Macular Corneal Dystrophy. Am J Ophthalmol 101(1):112-3 (1986).
5. Snip, RC., Kenyon, KR., Green, WR., Macular Corneal Dystrophy: Ultrastructural Pathology of Corneal Endothelium and Descemet's Membrane. Invest Ophthalmol 12(2):88-97 (1973).
6. Teng, CC., Macular Dystrophy of the Cornea: A Histochemical and Electron Microscopic Study. Am J Ophthalmol 62(3):436-54 (1966).
7. Klintworth, G. K., and Vogel, F. S. Macular corneal dystrophy. Am J Path.45:565 (1964)
8. Hassell, J.R., Newsome, D. A., Krachmer, J.H., Rodrigues, M.M. Macular corneal dystrophy: Failure to synthesize a mature keratan sulfate proteoglycan. Proc. Natl. Acad. Sci. USA. 77(6): 37-5-3709 (1908)
9. Thonar, EJ., Lenz, ME., Klintworth, GK., Caterson, B., Pachman, LM., Glickman, P., Katz, R., Huff, J., Kuettner, KE., Quantification of Keratan Sulfate in Blood as a Marker of Cartilage Catabolism. Arthritis Rheum 28(12):1367-76 (1985).
10. Cursiefen, C., Hofmann-Rummelt, C., Schlotzer-Schrehardt, U., Fischer, DC., Kuchle, M., Immunophenotype Classification of Macular Corneal Dystrophy: First Case Report of Immunophenotype IA Outside of Saudi Arabia, a Clinical Histopathological Correlation with Immunohistochemistry and Electron Microscopy. Klin Monatsbl Augenheilkd 217(2):118-26 (2000).
11. Klintworth, GK., Oshima, E., al-Rajhi, A., al-Saif, A., Thonar, EJ., Karcioglu, ZA., Macular Corneal Dystrophy in Saudi Arabia: A Study of 56 Cases and Recognition of a New Immunophenotype. Am J Ophthalmol 124(1):9-18 (1997).
12.Akama, TO., Nishida, K., Nakayama, J., Watanabe, H., Ozaki, K., Nakamura, T., Dota, A., Kawasaki, S., Inoue, Y., Maeda, N., Yamamoto, S., Fujiwara, T., Thonar, EJ., Shimomura, Y., Kinoshita, S., Tanigami, A., Fukuda, MN. Macular Corneal Dystrophy Type I and Type II are Caused by Distinct Mutations in a New Sulfotransferase Gene. Nat Genet 26(2):237-41 (2000).
13. Scott JE. Proteoglycan: collagen interactions and corneal ultrastucture. Biochem Soc Trans 1991;19:85-91.
14. Nakazawa, K., Hassell, JR., Hascall, VC., Lohmander, LS., Newsome, DA., Krachmer, J. Defective processing of keratan sulfate in macular corneal dystrophy. J Biol Chem. 1984 Nov 25;259(22):13751-7.
15. Akova, YA., Kirkness, CM., McCartney, AC., Ficker, LA., Rice, NS., Steele, AD., Recurrent Macular Corneal Dystrophy Following Penetrating Keratoplasty. Eye 4 ( Pt 5):698-705 (1990).
| |
|
|
|
|
|
|
 Welcome, please sign in
Welcome, please sign in