|
|
 |
|
|
|
|
|
Phenotypic Variations in X-linked Retinitis Pigmentosa
Digital Journal of Ophthalmology 1998
Volume 4, Number 1
August 1, 1998
|
Printer Friendly
|
S Andréasson | University of Lund, Sweden V Ponjavic | University of Lund, Sweden M Abrahamson | University of Lund, Sweden B Ehinger | University of Lund, Sweden W Wu | W.K. Kellogg Eye Center, University of Michigan R Fujita | W.K. Kellogg Eye Center, University of Michigan M Buraczynska | W.K. Kellogg Eye Center, University of Michigan A Swaroop | W.K. Kellogg Eye Center, University of Michigan
|
|
|
| Abstract | Objective
Clinical phenotypes were examined in Swedish families with x-linked retinitis pigmentosa caused by different mutations in the RPGR gene. The clinical findings in patients demonstrated a severe form of retinitis pigmentosa with visual handicap early in life. Furthermore, heterozygous carriers in these families revealed a wide spectrum of clinical features FROM minor symptoms to severe visual disability. These families SHOW a variable clinical phenotype resulting FROM different mutations in the RPGR gene.Keywords
X-Linked. Retinitis Pigmentosa, RPG-Gene | | | Introduction | | X-linked retinitis pigmentosa is a severe form of retinal degeneration, which manifests early symptoms of night blindness, visual field defects and decreased visual function. Recently, the gene RPGR (Retinitis Pigmentosa GTPase Regulator) had been cloned FROM the RP3 region, which seems to be the most common form of x-linked RP.[1-3] Different phenotypes in x-linked RP have been described earlier by many authors,[4-7] but until now, it has not been possible to correlate the phenotype to a precise gene mutation/genotype. This new knowledge requires further studies of patients with this disease. | |
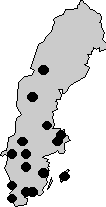
Figure 1
(Andréasson and associates). 15 families with x-linked retinitis pigmentosa FROM different parts of Sweden.
|
|
| Results | The two families with splice mutation in intron 10 and 13 demonstrated similar phenotypes with visual field constricted less than 50 degree with V4.e and less than 5 degree with I4e. The visual acuity was at least 20/50 besides the 76 years old man, who only had 20/100. The fundus SHOW similar features with narrow vessels, spicular pigmentary changes in all 4 quadrants. Full-field ERG recordings could be obtained and 30Hz flicker responses were measurable in all with at least 1 mVolt. In one young patient of 17 years with a splice mutation in intron 10 remaining rod responses could also be obtained (Table 1 and 2).
Most of the carriers had minimal symptoms. All carriers had some characteristic for the carrier state. Two obligate carriers had normal 30 Hz flicker implicit time but had pigmentation in the eyes.
The family with a deletion of exon 8-10 demonstrated a different phenotype. The two RP patients (age 34 and 41 years), were severely visually-handicapped. Full-field electroretinogram using the computer average technique and bandpass filter could not demonstrate any residual responses. The visual field were severely constricted. The fundus revealed narrow vessels, spicular pigmentation on all four quadrants and a pale optic disc (Table 3 and Figure 3).
The carriers in this family were severely affected. The visual field were constricted and full-field electroretinogram demonstrated markedly reduced responses for both rods and cones. The young carrier was at first examination 8 years old and was reexamined at age 15 (Table 3 and Figure 2). | |
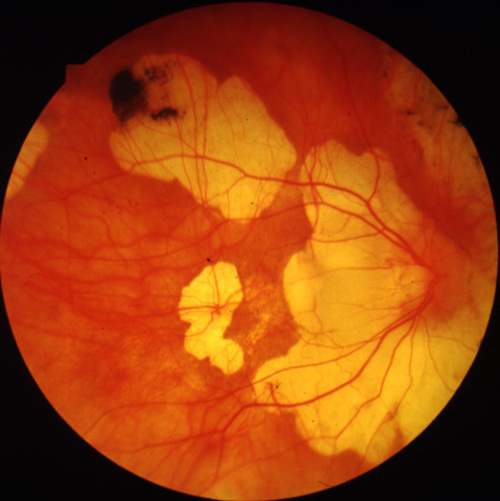
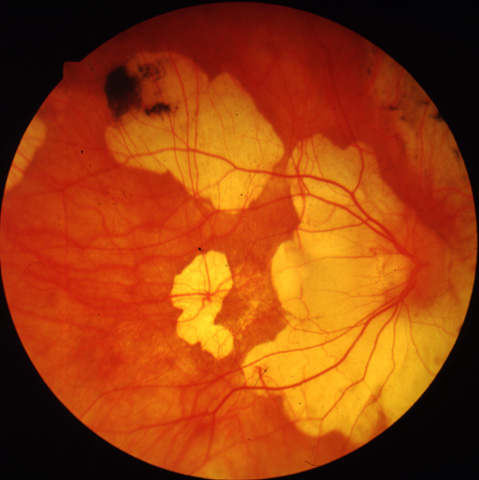
Figure 3a
Figures 3a-3c. (Andréasson and associates). Fundus photographs FROM three members in the family xlrp-126 with a microdeletion in the RPGR gene. Carrier I:1 age 66 (3a) demonstrated attenuated vessels, spicular pigment and atrophy of central fundus. Patient II:1 age 41 (3b) showed typical bone-spicular pigmentary retinal degeneration. Carrier III:1 age 8 (3c) had only minor retinal changes.
|
|

Figure 3b
|
|
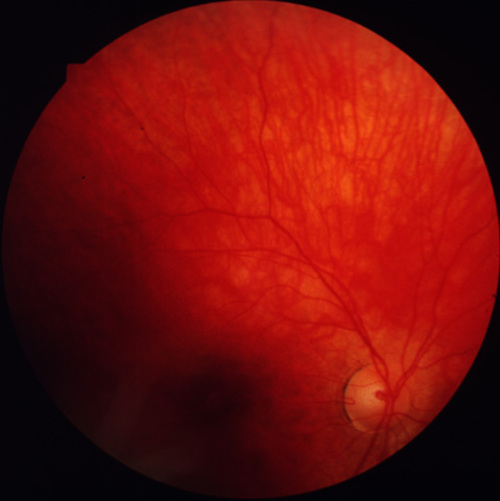
Figure 3c
|
|
| Discussion | X-LINKED RETINITIS PIGMENTOSA IN SWEDEN
During the last ten years we have continuously registered families with different forms of retinitis pigmentosa in the Swedish RP registry. This registry now contains almost 2,000 patients suffering FROM this disease, and we believe, that the registry includes 50-60 % of the patients in Sweden.
In several families with retinitis pigmentosa it is been difficult to clarify the x-linked trait. There are many isolated male RP patients or women with atypical form of RP, who are classified as undetermined or simplex. Recently, we identified about 100 patients in 15 families with x-linked retinitis pigmentosa. These 15 families live in different parts of Sweden (Figure 1) and have all been examined with full-field electroretinogram at the Departments of Ophthalmology, University hospital of Lund. All families have Swedish origin, and SHOW typical xlrp-trait with no male to male transmission and the women are less affected compared to the men in the same family. As part of molecular and genetic studies at the Kellogg Eye Center, we have determined the underlying defect in the RPGR gene in a number of XLRP families.[8,9] This report presents the clinical DATA FROM three of the Swedish families with X-linked RP.
PHENOTYPES ASSOCIATED WITH DIFFERENT MUTATIONS ON THE RPGR-GENE
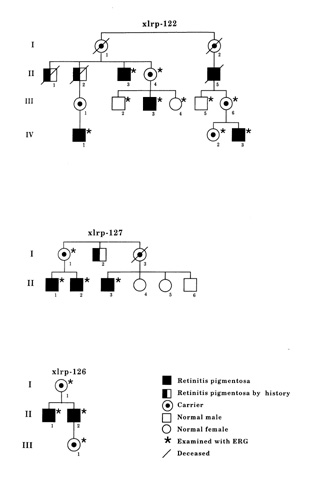
The pedigree of the three families are shown in Figure 2. Two of the families were carrying a splice defect in the RPGR -gene.[8] In family xlrp-127, there was an intron 13 splice defect, and in family xlrp-122, there was an intron 10 splice defect. The third family (xlrp-126) has a microdeletion spanning exons 8-10 of the RPGR gene.[9]
These families underwent ophthalmological examination included best corrected visual acuity, slit lamp inspection, ophthalmoscopy and fundus photography. Kinetic perimetry was performed with a Goldmann perimeter using standardized objects I4e and V4e. Full-field electroretinograms FROM all patients and carriers were recorded in a Nicolet Compact Four analysis system (Nicolet Biomedical Instruments, Madison, Wisconsin), as described previously.[10]
Most of these patients have been examined at least twice for followed up, which is demonstrated for one young carrier.
Conclusion
In Sweden with rather few inhabitants, 15 families with x-linked RP were identified and examined for mutations in the RPGR gene. By molecular genetics studies four different mutations in the RPGR-gene were identified. The phenotypes in three of these families were examined and it seems that these could be correlated to the mutations in the RPGR gene.
The families with a splice mutation in intron 10 and 13[8] were associated with severe phenotype of RP, but the examined patients retain useful vision even at the age of 40 and 50 yrs. In these families, the heterozygous carriers have minor symptoms and examination including full-field electroretinogram could only demonstrate minor changes.
In contrast, a family carrying a deletion of exons 8 -10[9], demonstrate a more severe disease with early visual impairment. The carriers in this family had also visually impairment with marked reduced retinal function measured by full-field electroretinograms.
We conclude that X-linked RP is, in general, associated with a more severe clinical phenotype, compared to other inherited types of retinitis pigmentosa. There is a variability of the phenotype depending on the nature of RPGR gene defect. Additional studies on other XLRP families with similar and different mutations in the RPGR gene will help in better understanding of the relationship between the genotype and the phenotype. [11,12] | |
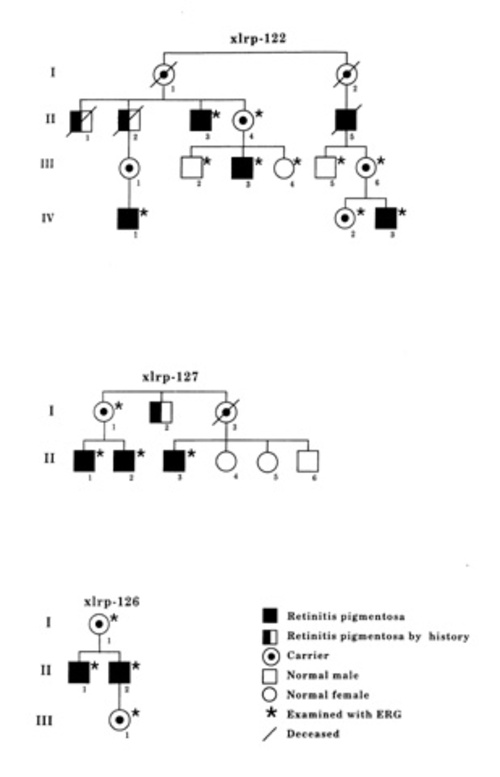
Figure 2
(Andréasson and associates). Pedigrees of the three Swedish families with x-linked retinitis pigmentosa.
|
|
| Acknowledgements | | Susanne Bauer, IngMarie Holst, Boel Nilssson Dept of Ophthalmology, University of Lund, Lund. This study was supported by grants FROM the The Swedish Society of Medicine, the National Institutes of Health (RO1 EY07961 and core grant EY07003), The Foundation Fighting Blindness, Hunt Valley, Md., and Research to prevent Blindness, New York. The research was carried out within research organizations sponsored by the RP Foundation. A.S. is recipient of a Research to Prevent Blindness Lew R. Wasserman Merit Award. | | | References | 1) Fujita R, Swaroop A: RPGR: Part one of the X-linked retinitis pigmentosa story. Mol Vis. 1996;2:4. URL: http://www.emory.edu/MOLECULAR_VISION/v2/fujita
2) Meindl A, Dry K, Herrmann K, Manson F, Ciccodicola A, Edgar A, Carvalho M.R.S, Achatz H, Hellebrand H, Lennon A, Migliaccio C, Porter K, Zrenner E, Bird A, Jay M, Lorenz B, Wittwer B, Dúrso M, Meitinger T, Wright A: A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nature Genet. 1996;13:35-42.
3) Roepman R, van Duijnhoven G, Rosenberg T, Pinckers AJLG, Bleeker-Wagemakers LM, Bergen AAB, Post J, Beck A, Reinhardt R, Ropers HH, Cremers FPM, Berger W: Positional cloning of the gene for X-linked retinitis pigmentosa 3: homology with the guanine-nucleotide-exchange factor RCC1. Hum Mol Genet. 1996;5:1035-1041.
4) Berson EL, Jakobiec F, Albert D: Principles and practice of Ophthalmology. Hereditary retinal diseases. Retinitis Pigmentosa and allied diseases. 1995;106.
5) Fishman, GA, Farber, M, Derlacki, DJ: X-linked retinitis pigmentosa: profile of clinical findings. Arch Ophthalmol. 1988;106:369-375.
6) Wright AF, Bhattacharya SS, Aldred MA, Jay M, Carothers AD; Thomas NS.Bird AC, Jay B; Evans HJ: Genetic localisation of RP2 type of X-linked retinitis pigmentosa in a large kindred. J Med Genet. 1991:28:453-457.
7) OMIM 312610. URL:http://www3.ncbi.nlm.nih.gov/Omim/
8) Fujita R, Buraczynska M, Gieser L, Wu W, Forsythe P, Abrahamson M, Jacobson SG, Andreasson S, Sieving PA, and Swaroop A: Analysis of the RPGR gene in 11 families with the RP3 genotype: Paucity of mutations in the coding region, but splice defects in two families. Am J Hum Genet 61:571-580, 1997.
9) Buraczynska M, Wu W, Fujita R, Buraczynska K, Phelps E, Andreasson S, Bennett J, Birch DG, Fishman GA, Hoffman DR, Jacobson SG, Musarella MA, Sieving PA, and Swaroop A: Spectrum of mutations in the RPGR gene identified in 20% of families with X-linked retinitis pigmentosa. Am J Hum Genet. in press.
10) Andréasson S, Ponjavic V, Ehinger B: Full-field electroretinogram in a patient with cutaneous melanoma-associated retinopathy. Acta Ophthalmol. 1993;71:487-90.
11) Jacobson SG, Buraczynska M, Milam AH, Chen C, Jarvalainen M, Fujita R, Wu W, Huang Y, Cideciyan AV, and Swaroop A: Disease expression in X-linked retinitis pigmentosa caused by a putative null mutation in the RPGR gene. Invest Ophthal Vis Sci 38:1983-1997, 1997.
12) Fishman GA, Grover S, Buraczynska M, Wu W, and Swaroop A: A new two base-pair deletion in the RPGR gene in an Afro-American family with X-linked retinitis pigmentosa. Arch Ophthalmol. in press. | | | Tables |
Table 1. Results FROM full-field electroretinograms in family 127
|
Age |
Blue flash
amplitude |
White flash
amplitude |
30 Hz flickering white flash amplitude |
30 Hz flickering white flash
implicit time |
|
yrs |
µV |
µV |
µV |
ms |
carrier (I:1) |
46 |
95 |
184 |
39 |
30,8 |
patient (II:1) |
27 |
0 |
5 |
1,4 |
38,8 |
patient (II:2) |
23 |
0 |
0 |
1,2 |
39,2 |
patient (II:3) |
25 |
0 |
7 |
3,2 |
40,8 |
|
|
|
|
|
|
Mean (± 2 SD)
in 70 controls |
|
166 ±115 |
315 ±196 |
63 ±41 |
29,3 ±3,3 |
Table 2. Results FROM full-field electroretinograms in family 122
|
Age |
Blue flash
amplitude |
White flash
amplitude |
30 Hz flickering white flash amplitude |
30 Hz flickering white flash
implicit time |
|
yrs |
µV |
µV |
µV |
ms |
carrier (II:4) |
69 |
100 |
234 |
30 |
31,2 |
carrier (III:6) |
44 |
82 |
195 |
42 |
35,2 |
carrier (IV:2) |
18 |
164 |
305 |
73 |
31,6 |
patient (II:3) |
76 |
0 |
0 |
1 |
62 |
patient (III:3) |
43 |
0 |
0 |
0,8 |
63 |
patient (IV:1) |
42 |
0 |
0 |
2,3 |
43 |
patient (IV:3) |
17 |
40 |
88 |
30 |
38,4 |
|
|
|
|
|
|
Mean (± 2 SD)
in 70 controls |
|
166 ±115 |
315 ±196 |
63 ±41 |
29,3 ±3,3 |
Table 3. Results FROM full-field electroretinograms in family 126
|
Age |
Blue flash
amplitude |
White flash
amplitude |
30 Hz flickering white flash amplitude |
30 Hz flickering white flash
implicit time |
|
yrs |
µV |
µV |
µV |
ms |
carrier (I:1) |
66 |
0 |
22 |
10 |
40 |
carrier (III:1) |
8 |
100 |
184 |
38 |
31,2 |
patient (II:1) |
41 |
0 |
0 |
0 |
|
patient (II:2) |
34 |
0 |
0 |
0 |
|
|
|
|
|
|
|
Mean (± 2 SD)
in 70 controls |
|
166 ±115 |
315 ±196 |
63 ±41 |
29,3 ±3,3 |
| |
|
|
|
|
|
|
 Welcome, please sign in
Welcome, please sign in