|
|
 |
|
|
|
|
|
Search for Correlations between Severity of Retinitis Pigmentosa and Primary Mutations
Digital Journal of Ophthalmology 1998
Volume 4, Number 8
October 15, 1998
|
Printer Friendly
|
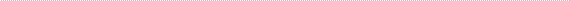
Eliot L. Berson, M.D. | Harvard Medical School, Massachusetts Eye and Ear Infirmary Michael A. Sandberg, PhD. | Harvard Medical School, Massachusetts Eye and Ear Infirmary Thaddeus P. Dryja, M.D. | Harvard Medical School, Massachusetts Eye and Ear Infirmary
|
|
|
| Introduction | Variability of clinical expression of retinitis pigmentosa has been described repeatedly over the last century but has remained an unexplained aspect of this GROUP of diseases [1–4]. Molecular genetic identification of mutant genes has provided new opportunities to EXPLAIN variable clinical expression as patients can now be subclassifed according to mutation.
We investigated to what extent the severity of retinitis pigmentosa is due to the domain altered by a given rhodopsin mutation [5,6]. We compared ocular function among 128 patients ages 7 to 73 with mutations altering either the intradiscal, transmembrane, or cytoplasmic domains of the rhodopsin molecule. Figure 1 shows a schematic model of human rhodopsin showing the seven transmembrane domains, the location of amino acid residues affected by dominant mutations causing retinitis pigmentosa, and the number of patients with each mutation included in this analysis [6].
We found that visual acuity, visual field diameter, dark-adapted sensitivity, and ERG amplitudes were highest for patients with mutations altering the intradiscal domain (or low-numbered codons) and lowest for patients with mutations altering the cytoplasmic domain (or higher-numbered codons). In the case of the ERG, mutations in the intradiscal domain resulted, on average, in ERG amplitudes that were 10- to 20-fold larger at a given age than mutations in the cytoplasmic domain (see TABLE 1) [6]. | |
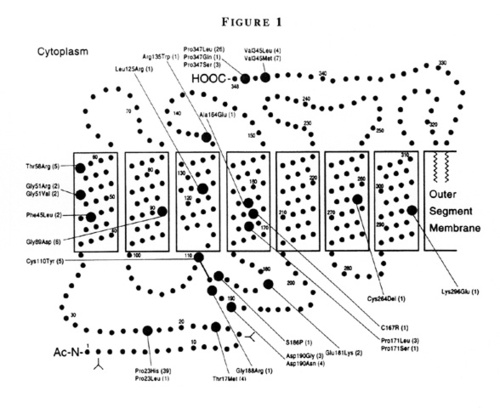
Figure 1
Schematic model of human rhodopsin showing the seven transmembrane domains, the locations of the amino acid residues affected by dominant mutations causing retinitis pigmentosa included in this report (large circles), and the number of patients with each mutation (parentheses). (From Sandberg MA, Weigel-DiFranco C, Dryja TP, Berson EL: Clinical expression correlates with location of rhodopsin mutation in dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 36:1934–1942, 1995.)
|
|
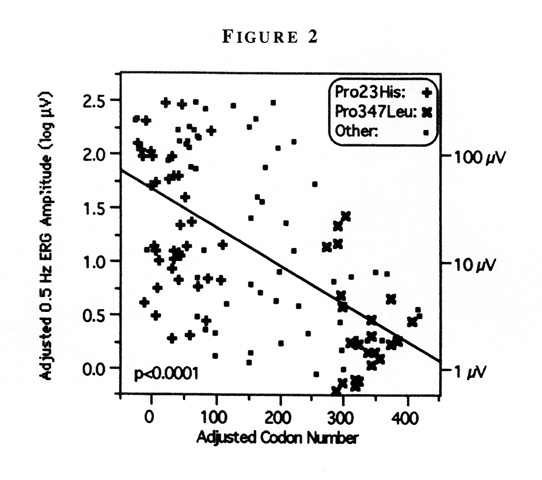
| Discussion | By multiple regression, all five measures of ocular function declined as codon number increased. The strongest relationship was seen between 0.5Hz ERG amplitude and codon number, as illustrated. In this plot, ERG amplitude decreased by 55% for every 100-unit increase in codon number (Figure 2) [6].
It is now known that patients FROM the same family or FROM different families with the same gene mutation can SHOW widely varying severity of disease at a given age. The first gene abnormality found to cause retinitis pigmentosa is a single-base substitution in codon 23 of the rhodopsin gene [7]. Although more than 70 mutations have been subsequently discovered in this gene, [8–14] this mutation (Pro23His) is the most common in the United States, accounting for approximately 7% of all cases of dominant retinitis pigmentosa [14]. The prevalence of this mutation in the United States provides an opportunity to use a relatively large sample size to study variability of clinical expression among patients with the same mutation.
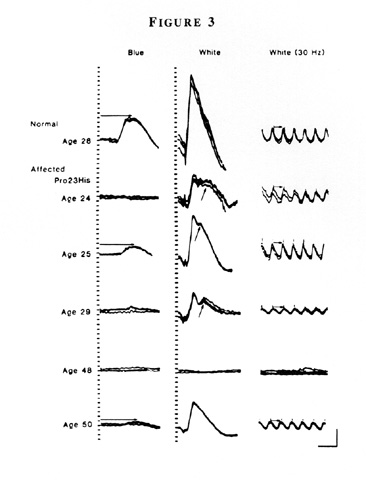
Representative ERGs are illustrated FROM a normal individual and 5 patients FROM a family with the Pro23His mutation (Figure 3).
Although all patients have reduced ERGs, considerable variability exists in the size of the responses at a given age. For example, an asymptomatic 50-year old patient has much larger responses than her night-deficient 48-year old sister. Fundus photographs FROM patients with the Pro23His mutation SHOW either the typical intraretinal pigment around the mid-periphery or little or no pigment. A 33-year old patient FROM one family shows more pigment than patients ages 45 and 48 FROM other families (Figure 4).
These ERGs and fundus photographs help substantiate that both intrafamilial and interfamilial clinical variability exists at a given age among patients with this mutation [15].
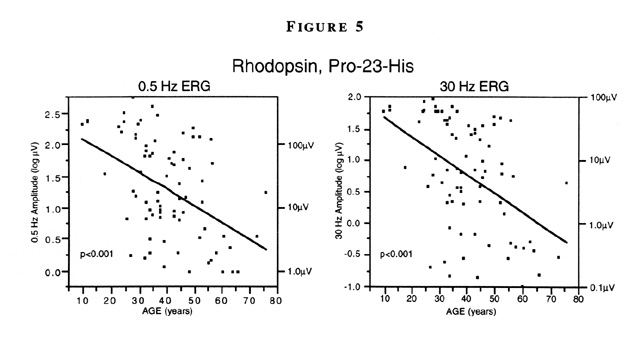
A cross-sectional analysis of patients with rhodopsin, Pro23His shows that both 0.5Hz and 30Hz ERG amplitude decline with increasing age. However, for patients within a given decade of age, 0.5Hz ERG amplitude can vary by as much as 100-fold and 30Hz ERG amplitude by as much as 1,000-fold (Figure 5) [16].
The marked variation in severity in patients with the same primary mutation must be due to environmental factors or modifier genes or both. We have been investigating the possible existence of a modifier gene. We have so far evaluated 15 families with 2 or more sibpairs with the Pro23His mutation. The 30Hz cone ERG was used as a measure of severity. ERG amplitude was corrected for age, sex, iris color, and refractive error.
We found that the distribution of patients when considered individually by 30Hz cone ERG amplitude appeared to be unimodal (Figure 6).
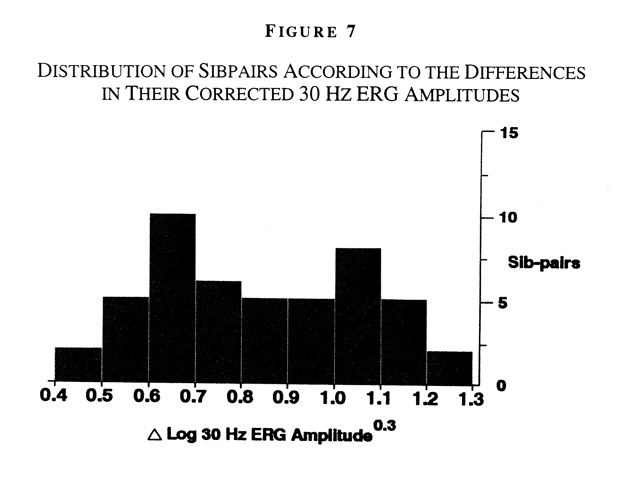
This is not evidence for or against the existence of a modifier gene; environmental factors or the effects of many modifier genes could be obscuring a bimodal distribution caused by a modifier gene. We then confined our analysis to affected siblings, as siblings share many environmental factors, thereby making the effects of a major modifier gene more pronounced. We calculated the absolute value of the difference in log10 30Hz ERG amplitude for each sibpair. We examined the distribution of the sibpairs according to these differences to see if they fit a normal distribution. A histogram of sibpairs with the Pro23His mutation, ranked according to the difference in log10 30Hz ERG amplitude for each pair, is illustrated (Figure 7).
Raw differences were raised to the 0.3 power to OPTIMIZE normalization. We found that the distribution of sibpairs did not fit a normal distribution, but in fact appeared to be bimodal [16].
The technique of Bhattacharya [17] as described by Everitt and Hand [18] was used to resolve the difference of sibpairs INTO Gaussian components. This procedure allowed us to estimate the means and standard deviations of 2 normal distributions within the histogram of 30 Hz ERG amplitude differences. The 2 distributions were interpreted to signify 2 groups of sibpairs. One category of sibpairs, designated as "sims" or similar sibs, had a mean difference of cone-related severity of about 0.3 log10 units. The second category of sibpairs, designated as "contras" or contrasting sibs, had a difference of about 0.93 log10 units. Stated in another way, the average pair of sibs in the sim GROUP had a 2-fold difference in severity as measured by the 30 Hz cone ERG (i.e., 10 to the 0.3 power equals 2), while the average pair of sibs in the contra GROUP had an 8.5-fold difference in severity (i.e., 10 to the 0.93 power equals 8.5). The difference in the means between the sims and the contras is about 0.63 log10 units, suggesting that the presence or absence of a modifier gene accounts for a 4.3-fold difference in severity (i.e., 10 to the 0.63 power equals 4.3) [16]. More families are under study to confirm this distribution and confirm the magnitude of this effect.
The existence of a bimodal distribution of sibpairs is a strong indicator that a gene is responsible for at least some of the variation in severity seen in this form of retinitis pigmentosa. Our GROUP is beginning a linkage study to search for the chromosomal location of this major modifier gene. Discovery of a modifier gene or genes might provide new insight INTO possible therapeutic modalities to slow the progression of this condition. However, based on our analyses to date, a major modifier gene may EXPLAIN only a portion of the variation in severity among patients with the Pro23His mutation. We are therefore also searching for additional factors which affect clinical expression of the disease. For example, we are evaluating the effect of various dietary constituents on the course of the disease with the same mutation.
Our experience with patients with Pro23His suggests that large datasets are required to perform these risk factor analyses. In our experience, the large range in severity of disease among patients with the same mutation is not specific to Pro23His. Because of this large variation among patients with the same primary mutation, clinicians and molecular geneticists should exercise caution before labelling a given mutation as "mild" or "severe." Such designation may not only be inaccurate, but may produce unwarranted assurance or anxiety in patients who read the scientific literature. Every effort should be made to obtain DNA and comprehensive clinical evaluations FROM large numbers of patients with a given mutation, selected with as little ascertainment bias as possible, before conclusions are made about the severity of disease associated with that mutation.
In summary, marked variability in severity of RP exists among patients with dominant rhodopsin mutations. Part of this variability relates to some mutations causing more severe disease on average than others. A general correlation exists between severity of disease and location of the affected amino acid in the polypeptide chain. Variability in clinical expression also exists among patients with the same primary mutation (e.g., rhodopsin, Pro23His). Part of this variation in severity appears due to a modifier gene; the remainder is possibly due to other modifier genes and/or environmental or dietary factors. | |

Figure 2
Schematic model of human rhodopsin showing the seven transmembrane domains, the locations of the amino acid residues affected by dominant mutations causing retinitis pigmentosa included in this report (large circles), and the number of patients with each mutation (parentheses). (From Sandberg MA, Weigel-DiFranco C, Dryja TP, Berson EL: Clinical expression correlates with location of rhodopsin mutation in dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 36:1934–1942, 1995.)
|
|
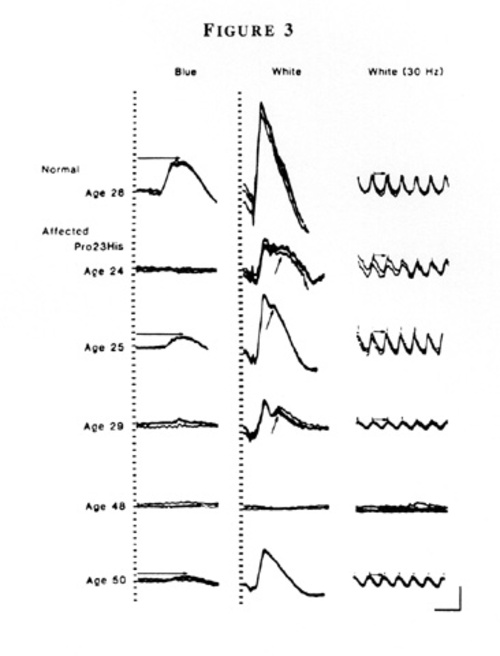
Figure 3
Full-field ERGs FROM an unaffected patient and five affected relatives in family 5850 with dominant retinitis pigmentosa and rhodopsin, Pro23His. Horizontal arrows in the left column designate rod b-wave implicit times, and in the right column, cone b-wave implicit times, i.e., time interval between stimulus flash and corresponding cornea-positive peaks. Oblique arrows in the middle column designate delayed rod-dominated peaks. Calibration symbol (lower right) designates 50 milliseconds horizontally and 100 µV vertically. (From Berson EL, Rosner B, Sandberg MA, and Dryja TP: Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (Pro23His) Arch Ophthalmol 109:92–101, 1991. Copyright 1991, American Medical Association.)
|
|
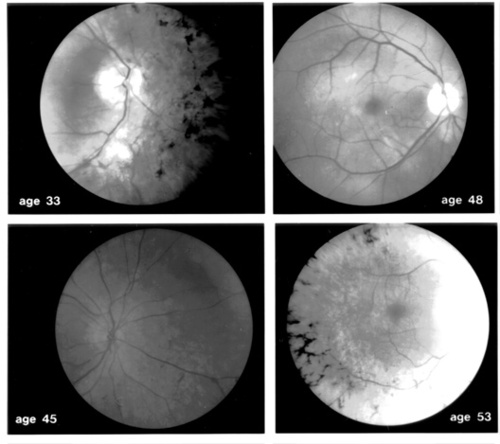
Figure 4
Representative fundus photographs FROM four patients with autosomal dominant retinitis pigmentosa with a C to A transversion in codon 23 of the rhodopsin gene to SHOW variability with respect to the extent of intraretinal bone spicule pigmentation among patients with the same gene defect. (From Berson EL: Ocular findings in a form of retinitis pigmentosa with a rhodopsin gene defect. Trans Amer Ophthalmol Soc 88:355–388, 1990.)
|
|

Figure 5
Full-field ERG amplitudes to 0.5 Hz (left) and 30 Hz white light (right) as a function of age among patients with rhodopsin, Pro23His. In each case the solid line represents the least-square fit of ERG amplitude versus age; regression of ERG amplitude on age was significant for each test condition (p<0.001). Within 10-year intervals amplitudes can vary by 100- to 1,000-fold.
|
|

Figure 6
Distribution of Patients by 30 Hz ERG Amplitudes Corrected for Sex, Age, Iris Color, and Refractive Error. Histogram of log10 30 Hz cone ERG amplitudes (corrected for sex, age, refractive error, and iris color) FROM patients with the rhodopsin, Pro23His mutation. The distribution appears unimodal.
|
|

Figure 7
Distribution of Sibpairs According to the Differences in Their Corrected 30 Hz ERG Amplitudes. Histogram of log10 30 Hz cone ERG amplitude (corrected for sex, age, refractive error, and iris color) FROM sibpairs with the rhodopsin, Pro23His mutation, ranked according to the difference within each pair. Raw differences were raised to the 0.3 power to OPTIMIZE normalization. The distribution appears bimodal. More families are under study to confirm this distribution.
|
|
| Acknowledgements | | This research was supported by grants EY00169 and EY08683 FROM the National Eye Institute, Bethesda, MD and by the Foundation Fighting Blindness, Baltimore MD. | | | References | 1. Leber, T., Die Pigmentdegeneration der Netzhaut und mit ihr verwandte Erkränkungen In: Graefe A, Saemisch-Hess T, eds. Handbuch der gesammten Augenheilkunde. 2nd ed., vol 7. Leipzig, Germany: W. Engelmann, pp 1076–1225 (1916)
2. Bell, J., Retinitis pigmentosa and allied diseases of the eye. In: Pearson, K., ed. Treasury of Human Inheritance. Vol 2. London: Cambridge University Press, pp 1–28 (1922)
3. Heckenlively, JR., Retinitis Pigmentosa. Philadelphia: J.B. Lippincott Co. (1988)
4. Berson, EL., Retinitis pigmentosa: The Friedenwald lecture. Investigative Ophthalmology and Visual Science 34:1659–1676 (1993)
5. Berson, EL., Rosner, B., Sandberg, MA., Weigel-DiFranco, C., Dryja, TP., Ocular findings in patients with autosomal dominant retinitis pigmentosa and rhodopsin, proline-347-leucine. American Journal of Ophthalmology 111:614–623 (1991)
6. Sandberg, MA., Weigel-DiFranco, C., Dryja, TP., Berson, EL., Clinical expression correlates with location of rhodopsin mutation in dominant retinitis pigmentosa. Investigative Ophthalmology and Visual Science 36:1934–1942 (1995)
7. Dryja, TP., McGee, TL., Reichel, E., et al., A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 343:364–366 (1990)
8. Sung, CH., Davenport, CM., Hennessey, JC. et al., Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proceedings of the National Academy of Science U.S.A. 88:6481–6485 (1991)
9. Dryja, TP., Hahn, LB., Cowley, GS., et al., Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proceedings of the National Academy of Science U.S.A. 88:9370–9374 (1991)
10. Sheffield, VC., Fishman, GA., Beck, JS., et al., Identification of novel rhodopsin mutations associated with retinitis pigmentosa by GC-clamped denaturing gradient gel electrophoresis. American Journal of Human Genetics 49:699–706 (1991)
11. Inglehearn, CF., Keen, TU., Bashir, R., et al: A completed screen for mutations of the rhodpsin gene in a panel of patients with autosomal dominant retinitis pigmentosa. Human Molecular Genetics 1:41–45 (1992)
12. Bunge, S., Wedemann, H., David, D., et al. Molecular analysis and genetic mapping of the rhodopsin gene in families with autosomal dominant retinitis pigmentosa. Genomics 17:230–233 (1993)
13. Sung, CH., Davenport, CM., Nathans J., Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa. Journal of Biological Chemistry 268:26645–26649 (1993)
14. Vaithinathan, R., Berson, EL., Dryja, TP., Further screening of the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. Genomics 21:461–463 (1994)
15. Berson, EL., Rosner, B., Sandberg, MA., Dryja, TP., Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (Pro23His). Archives of Ophthalmology 19:92–101 (1991)
16. Dryja, TP., Sandberg, MA., Ott, J., Berson, EL., Evidence for a genetic locus modulating the severity of retinitis pigmentosa caused by the rhodopsin mutation P23H. Investigative Ophthalmology and Visual Science 38(suppl):797 (1997)
17. Bhattacharya, CG., A simple method for resolution of a distribution of Gaussian components. Biometrics 23:115–135 (1967)
18. Everitt, BS., Hand, DJ., Finite mixture distributions. London, Chapman & Hall, pp 49–51 (1981) | | | Tables |
Table 1: Ocular Function Versus Domain Altered by
a Rhodopsin Mutation in Patients with Dominant Retinitis Pigmentosa
Parameter |
Intradiscal |
Transmembrane |
Cytoplasmatic |
P-value|| |
|---|
Visual acuity* |
0.31±0.10 (60)
[20/24] |
-0.19±0.16 (26)
[20/30] |
-0.40±0.13 (42)
[20/34] |
0.0001 |
Visual field† |
91.6±4.4 (60) |
81.1±6.6 (26) |
64.4±5.3 (41) |
0.0007 |
Dark-adapted sensitivity‡ |
-1.40±0.12 (54) |
-2.10±0.18 (25) |
-3.26±0.15 (36) |
<0.0001 |
ERG amplitude (0.5 Hz)§ |
1.48±0.09 (59)
[30.2µV] |
1.36±0.14 (24)
[22.9µV] |
0.39±0.11 (39)
[2.5µV] |
<0.0001 |
ERG amplitude (30 Hz)§ |
1.00±0.09 (59)
[10.1 µV] |
0.76±0.15 (24)
[5.8 µV] |
-0.05±0.12 (39)
[0.9 µV] |
<0.0001 |
Values are mean±SE (number of patients)
ERG=electroretinogram
* Visual acuity normalizied after conversion to ranks, adjusted for age;
visual acuity corresponding to mean value in brackets.
† Diameter (degrees), adjusted for age
‡ Log sensitivity relative to normal, adjusted to age.
§ Log amplitude (µV), adjusted for age and equivalent sphere;
geometric mean amplitude in brackets (normal, ³
350µV to 0.5 Hz flashes and ³
50µV to 30 Hz flashes).
|| Based on multiple regression.
(From Sandberg MA, Weigel-DiFranco C, Dryja TP, Berson EL: Clinical expression
correlates with location of rhodopsin mutation in dominant retinitis pigmentosa.
Invest Ophthalmol Vis Sci 36:1934–1942, 1995.)
| |
|
|
|
|
|
|
 Welcome, please sign in
Welcome, please sign in