|
|
 |
|
|
|
|
|
Phenotypic Variation in Patients with Mutation in the Peripherin/RDS Gene
Digital Journal of Ophthalmology 1999
Volume 5, Number 2
May 1, 1999
|
Printer Friendly
|
|
|
|
|
| Abstract | Objective
Peripherin/rds is a transmembrane glycoprotein that structurally helps form and stabilize photoreceptor disks in rods and cones. Peripherin/rds and a homologous protein, rom-1, are present within the disk rims of rods, and probably also cones, as covalently bound homodimers that form a tetramer with one another through noncovalent bonds. This tetramer, possibly combined with other rim proteins, may represent the margin template of Corless and Fetter, which when aligned in the disc rim region, forms the terminal loop complex, which appears to be the primary morphogen for disk formation. The peripherin/rds -rom-1 complex may also interact with cytoskeletal proteins to help align and stabilize the margins of the disk rims and the incisures. Mutations of peripherin/RDS have been associated with several forms of retinal and macular degeneration, including autosomal dominant retinitis pigmentosa, digenic retinitis pigmentosa (with ROM1 mutations), butterfly or pattern-like macular dystrophy, fundus flavimaculatus, cone-rod dystrophy, and various forms of regional and diffuse choroidal atrophy. Missense mutations may interfere with the interactions of peripherin/RDS with other rod or cone specific proteins. Such mutations may result in varying phenotypes either directly by causing photoreceptor death (producing retinitis pigmentosa or cone dystrophy) or by leading to excessive shedding of abnormal disks with progressive accumulation of lipofuscin within retinal pigment epithelium (leading to macular disease). Null mutations produce a slow, late-onset macular/retinal degeneration suggesting that the production of gene product FROM only one normal allele is insufficient to maintain normal disk renewal for a lifetime.Keywords
Humans, retina, retinal degeneration, peripherin gene | | | Introduction | Peripherin/rds is a 39 kd glycoprotein that is membrane bound and found exclusively within the disks of the outer segments of rods and cones [1-3]. The gene resides on mouse chromosome 17 and on the short arm of human chromosome 6; in both species the gene codes for a 346 amino acid product [4]. Peripherin/rds is composed of four putative membrane-spanning domains with both the amino-terminal and the carboxy-terminal ends within the cytoplasm (Figure 1) [4,5].
The larger of two intradiskal loops, D2, has a conserved glycosylation site that may form homophilic bonds across the disk space, contributing to the bend of the edge of rod and cone disks [2]. Two peripherin/rds protein molecules bind covalently to create homodimers (Figure 2).
In rods, these peripherin/rds homodimers develop non-covalent bonds with covalently bonded homodimers composed of another rod-specific glycoprotein rom-1 to form tetramers [6-8]. In cones, homodimers of peripherin/rds appear to form tetramers with homodimers of either rom-1 or a homolog [9]. These tetramers appear to play an integral role in the development and stability of disks. Mutations of peripherin/RDS produce retinal degeneration both in man and in animals presumably by interfering with these functions.
Retinal Degeneration Slow (rds)
Much of what we understand to be the role of peripherin/RDS in the photoreceptors stems FROM the study of the naturally occurring mouse model termed retinal degeneration slow, or rds. This strain, which was first described in 1978 by Van Nie et al. [10], has a 10 kbp insertion in codon 230 creating, in effect, a null allele [11-13]. The disease in mice has a very different phenotype depending on whether the mutation allele is present in the homozygous or heterozygous state. In the homozygous state no peripherin is present, normal rod or cone disks are absent FROM birth, and a severe retinal degeneration ensues [14]. In animals that are heterozygous, rod and cone disks are formed at birth but are distorted and ballooned because of apparent lack of normal disk morphogenesis and disk stability [15]. This lack of the normal formation and stability of disks in the rds heterozygous mouse is due to haploinsufficiency, which is defined as the inability of only one allele to produce enough gene product for a given process or function, in this case, the formation of normal photoreceptor outer segment disks [16].
Photoreceptor Disk Morphogenesis
Steinberg proposed the model whereby photoreceptor disks form as successive "shelf-like" evaginations FROM the plasma membrane in the region of the inner face of the connecting cilium [17]. The narrow extracellular space between these shelves represents the future intradiskal space. By a process of differential membrane growth and fusion, the membranes of adjacent shelves become the disks of the photoreceptors outer segments.
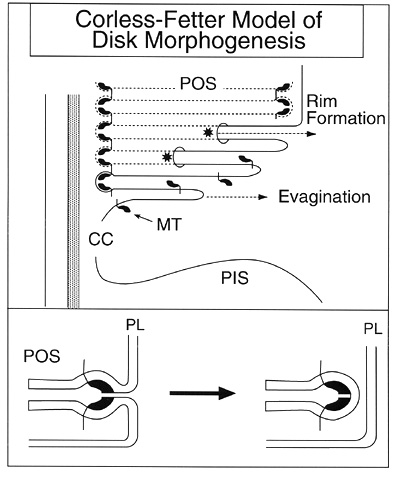
According to Corless and Fetter, photoreceptor disk formation and the closure of the disk rims require a separate mechanism FROM that which produces the shelf-like evaginations (Figure 3).
The creation of the disk perimeter involves the organization of a terminal loop complex formed FROM margin templates, which are semilunar or crescent-shaped subunits that must come together before the rims of disks can be formed [18-20].
The process of photoreceptor disk morphogenesis begins when these subunits of rim-specific protein complexes, called extracellular margin templates, are attached to the vitreal base of the photoreceptor outer segment plasma membrane in the region of the connecting cilium (Figure 3) [20]. Corless and Fetter hypothesized correctly that the margin templates would be transmembrane proteins with both cytoplasmic and extracellular domains. The extracellular domains interact across the nascent disk space to bend the shelf membrane INTO the rim structure.
The cytoplasmic domains of the margin templates attach to filaments that connect the disk rims to each other, thus aligning the edges of the disks. Attachments also form FROM these cytoplasmic filaments to the cytoskeletal network connecting the disk rims to the inner aspect of the plasma membrane, thus providing stability to the photoreceptor outer segments.
Additional extracellular margin templates are attached to both the scleral and vitreal surfaces of the first shelf-like evagination of the plasma membrane at the proximal end of the photoreceptors in the region of the axoneme. When the second evagination occurs, formation of a nascent disk begins by a progressive circumferential fusion of the scleral and vitreal surfaces of the shelf membrane at the rim. This fusion is driven by the same template-driven process that depends on the aligning of the crescent-shaped margin templates to create the terminal loop complexes within the concave extracellular space between the two shelves.
The template created by the terminal loop complex forces the curvature of the membrane to form the disk edge and promotes the elongation of the membrane of the disk rim. In this fashion, the disk margin template propagates the formation of a disk edge, termed the growth point, which lies inside and will soon separate FROM the plasmalemma. This leading edge of disk closure progressively extends circumferentially in both directions eventually fusing on the side of the outer segment farthest FROM the cilium to completely enclose the disks within the outer plasma membrane. Although the disks in rods become completely separated FROM the plasma membrane, the process for cones fails to extend 360 degrees to completely enclose off the disks and, for a good portion of the cone outer segments, the disks remain open to the extracellular space. Shedding of photoreceptor outer segments may involve a mechanism similar to the reverse of disk morphogenesis, where disks become exposed to the extracellular space facilitating phagocytosis of outer segments by retinal pigment epithelium.
As indicated above, both the peripherin/rds and rom-1 homodimers form a non-covalently bound tetradimer (Figure 2), which may represent the subunit Corless and Fetter term the extracellular marginal template. The intradiskal loops of the peripherin/rds -rom-1 tetradimers may participate in additional non-covalent bonding at the intradiskal margins. The tertiary structures produced by these tetramers may represent the fully assembled terminal loop complexes. The normal development of photoreceptor disks requires exact, or stoichiometric concentrations of both extracellular marginal templates and fully aligned terminal loop complexes. If too few of these templates and their associated complexes are formed, for example when too little gene product for peripherin/rds is available, the proper shape and enclosure for the photoreceptor disks will not occur, and the unchecked growth of the shelf surfaces will produce ballooned, distorted disk membranes in the shape of whorls [16]. Missense mutations affecting peripherin/rds or rom-1 could create changes in the tertiary structures of terminal complexes that lessen their efficiency in performing the template function or result in formation of atypical rds-rom-1 heterodimers. Depending on whether the mutation influenced to a greater degree a specific type of photoreceptor or the outer segment shedding process itself, the phenotype could be either a diffuse rod dystrophy, such as retinitis pigmentosa, a cone degeneration, or a macular dystrophy. | |

Figure 1
Secondary structure of the peripherin/RDS gene showing the cytoplasmic amino terminus and carboxy terminus and the large intradiskal loop that may play a role in interactions that bind together Rds:Rom-1 tetramers INTO the terminal loop complexes. The codons are designated by their encoded amino acids. Codons that are mutated in human disease are shown with white letters on colored circles according to the predominant phenotype for that mutation. All of the missense mutations within the D2 loop that are associated with either retinitis pigmentosa or cone/cone--rod dystrophy involve amino acid residues that are conserved in rom-1. The codon 25 2-bp deletion is listed as a null mutation but the phenotype was reported as retinitis punctata albescens. Three presumably null mutations (codon 140 4-bp ins, which causes a frame shift creating a stop mutation 8 aberrant amino acids later, the 2-bp deletion spanning codons 299 and 300, which creates a stop mutation 23 residues later, and the Tyr258ter mutation) are indicated in yellow because the phenotype appears only to involve the macula. (Modified FROM an illustration provided by Edwin M. Stone, M.D., Ph.D., and Kimberlie Vandenburgh, B.S.)
|
|
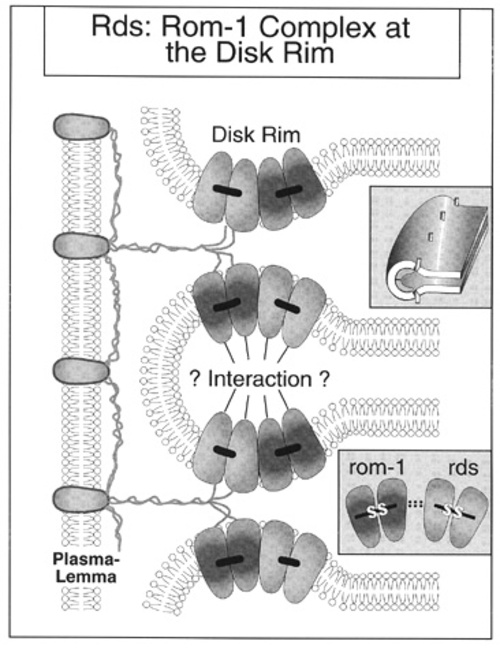
Figure 2
Rds:Rom-1 interactions at the disk rim, showing the homodimers of rds and rom-1 forming tetramers by non-covalent bonds. These tetramers, which may represent the extracellular margin templates of Corless and Fetter, may interact across the intradiskal space at the rim margin to form the terminal loop complex (upper insert). (Adapted FROM an illustration provided by Drs. G. Clarke and R. McInnis.)
|
|

Figure 3
Schematic illustration of the template theory of disk morphogenesis proposed by Corless and Fetter. Disk morphogenesis begins as a plate-like evagination FROM the plasma membrane in the region of the connecting cilium. The extracellular margin templates (MT) become aligned and assemble to form the terminal loop complexes that Corless and Fetter believe represent the primary morphogen of the disk rims. The asterisk denotes the leading edge of disk rim closure. CC is connecting cilium, POS is photoreceptor outer segment, PIS is photoreceptor inner segment, and PL is plasmalemma. (Adapted FROM Arikawa et al., 1992, and Corless and Fetter, 1987).
|
|
| Discussion | Phenotypes Associated with Mutations of Peripherin/RDS
Dominant Retinitis Pigmentosa
Kajiwara in 1991 first reported autosomal dominant retinitis pigmentosa (adRP) associated with mutations (Pro219del and Pro216Leu) of the peripherin/RDS gene [21]. Farrar et al in 1991 mapped a large family with autosomal dominant retinitis pigmentosa to the long arm of chromosome 6 [22]. That same year, this GROUP reported a 3-bp deletion (Cys118 or Cys119) of the RDS gene in this same kindred [23]. Wells et al. also reported the same C118(119)del associated with retinitis pigmentosa [24]. Kajiwara et al in 1993 reported the finding of 3 other mutations (Gly68Arg, Leu126Arg, Gly266Asp) associated with adRP [25]. In 1994 Kemp et al. reported Pro210Ser associated with adRP [26]. The stretch of 10 amino acids beginning at codon 210 appears to be important for retinal function because at least 8 different mutations within this range (Pro210Arg, Pro210Ser, Phe211Leu, Ser212Gly, Cys214Ser, Pro216Ser, Pro216Leu, Pro219del) have been associated with retinal degeneration [21,26-31]. Virtually all of the mutations within the D2 loop that are associated with the RP phenotype involve amino acid residues that are conserved in human rom-1 [12]. This suggests that the D2 loop, particularly the region FROM codon 210 to 219, may be an important site for interaction between rds and rom-1 homodimers. Kikawa in 1994 reported a Asn244Lys mutation of RDS associated with adRP with adRP with a prominent bull's eye maculopathy and rod-cone ERG loss [32,33]. This same codon, when changed to a histidine, has been reported with a cone-rod dystrophy, also with a bull's eye maculopathy [34]. Apfelstedt-Sylla et al. reported a 1 bp del in codon 307 associated with a late-onset diffuse form of adRP characterized by a widespread choriocapillaris atrophy component [35].
Retinitis punctata albescens
This phenotype has been reported in a heterozygous 2-bp deletion in codon 25 termed Trp25(2-bp del). The 59-year old male patient had advanced retinitis pigmentosa, involving the macula and with peripheral yellow-white subretinal flecks [36]. A 33-year old asymptomatic daughter had subretinal flecks, abnormal retinal vessels, and an abnormal ERG. Since this deletion creates a frame shift with a subsequent stop mutation 54 basepairs later, this mutation represents a null allele. The mechanism of disease is presumably by haploinsufficiency.
Digenic Retinitis Pigmentosa
Although originally reported to be associated with autosomal dominant retinitis pigmentosa [21], the Leu185Pro mutation of peripherin/RDS was demonstrated in 1994 to be associated with retinitis pigmentosa only when inherited in conjunction with either the ROM1 mutation Gly80(1-bp ins) or Leu114(1-bp ins), both of which represent null alleles [37]. This form of digenic inheritance is the simplest form of polygenic inheritance. The validity of this concept has been shown by the creation of the digenic genotype (Rom1+/-, Rds +/-, tgL185P) FROM breeding knock-out mice for the rom-1 gene (Rom1-/-) to transgenic mice that have one normal rds allele and a trangene for Leu185Pro (Rds +/-, tgL185P) [38]. The digenic animals, who were heterozygous for both the rom-1 null allele and who carried the L185P transgene on a +/- rds background, showed major rod outer segment abnormalities whereas animals that inherited either the L185P Rds transgene or the null allele for Rom1 alone did not.
Macular Dystrophy
Wells et al in 1993 reported patients with macular dystrophy in association with both Arg172Gln or Arg172Trp missense mutations of peripherin/RDS [24]. The phenotype in Arg172Trp appears to be the most completely characterized of the peripherin/RDS missense mutations [39-43]. The disease can present in adolescents as central pigmentary alterations with drusen-like deposits developing in the late teens and twenties. Later in the course of the disease focal and then central geographic choroidal atrophy occurs with, by at least the fifth decade, further loss of central visual acuity [42]. Late in the course of disease, the fundus appearance with the codon 172 mutation may simulate that of central areolar choroidal dystrophy. Most reports found greater cone than rod dysfunction evident on Ganzfeld ERG testing [24,39,41,42].
Butterfly or pattern-like macular dystrophy has been reported with Gly167Asp and Arg172Gly missense mutation, a 2-bp deletion beginning at last bp in codon 299, and a large deletion involving exons 2 and 3 of RDS [26,44-47]. Other mutations that have been associated with adult onset foveomacular dystrophy, which is similar to pattern dystrophy, are Pro210Arg [7,48] and 4-bp insertion of codon 140 [49,50]. A nonsense mutation (Tyr258ter) was also reported in association with a vitelliform macular dystrophy by this same GROUP [24].
Patients with null alleles of RDS appear to often involve the macula relatively early in the course of disease but this phenotype is believed to result FROM haploinsufficiency. Thus, greater understanding of the role of peripherin/rds may result FROM the knowledge of how mutations that presumably still code for a protein cause disease. Two of the RDS missense mutations occurring within the D2 loop that are associated with only the macular dystrophy phenotype alter amino acid residues (Arg142 and Arg172) that appear not to be conserved in human rom-1 [12]. As mentioned above, this differs FROM those missense mutations within the D2 loop that are associated with retinitis pigmentosa, where virtually all such mutations involve amino acid residues conserved with rom-1.
Fundus Flavimaculatus
A fundus flavimaculatus-like phenotype has been reported in two family members with a 3-bp deletion removing either Lys153 or Lys154 and in association with a null allele Arg46stop [35,51-53].
Cone or Cone-rod Dystrophy
Recently Fishman et al. described a family with an apparent cone dystrophy associated with a Ser27Phe mutation [54]. Cone-rod retinal dystrophy, often in association with a bull's eye maculopathy, has been associated with mutations Tyr184Ser, Asn244His, and Val200Glu [34,55]. For the codon 184, 244, and 200 mutations, but not the codon 27 mutation, the amino acid residue involved is conserved in rom-1 [12].
Choroidal Atrophy
Regional: Central areolar choroidal dystrophy
The fundus appearance of central areolar choroidal dystrophy has been reported in the late stage of macular dystrophy in several missense mutations, including Arg 142Trp [56], Arg172Trp [24,39-43], and a 2-bp deletion involving codon 193 creating a frame shift that ends in a termination codon 24 incorrect amino acid residues later [57].
Diffuse: Diffuse choriocapillaris atrophy
Diffuse choriocapillaris atrophy was reported in a 63-year-old woman with a mutation that involved a 8-bp insertion for a 5-bp deletion in codons 67 through 69, resulting in two amino acid changes, Met67Arg and Gly68His, as well as an arginine insertion at position 69 [57].
Mutations Associated with Multiple Phenotypes
A family was reported in 1993 with several members segregating for a 3-bp deletion of either Lys153 or Lys154 [51]. Central choriocapillaris atrophy was a prominent feature of the proband, a 75-year-old woman with adult-onset RP. Other members of this family had either pattern-like macular dystrophy or a fundus flavimaculatus phenotype. Gorin et al. reported several phenotypes associated with a Pro210Arg mutation, including age-related macular degeneration, retinitis pigmentosa, Stargardt disease, adult vitelliform degeneration, dominant drusen, and pattern dystrophy [27]. Fishman et al. reported both retinitis pigmentosa and pattern dystrophy within the same family in association with Pro216Ser [31]. The status of Pro216Ser as a disease-causing mutation is clouded slightly by the family reported by Richards et al. where 4 members had features of RP and pattern dystrophy, but 4 other relatives had the mutation and were normal on fundus examination [58]. The authors attributed this to reduced penetrance of the gene.
Phenotype of Null Allele (Haploinsufficiency)
Several mutations have been reported that either directly or through a frame shift result in a stop codon creating in essence a null allele. The first null allele reported, a 2-bp deletion involving codon 25 that creates a frameshift and a premature stop at codon 43, was described in association with what the authors termed retinitis punctata albescens [36]. Since the characteristic feature was numerous yellow-white subretinal flecks, others might call this phenotype fundus flavimaculatus.
The most common of the rds null alleles is Arg46stop, which has been reported in three families [35,52,53]. Apfelstedt-Sylla et al. consider the most appropriate descriptive term for the phenotype of their large family, which had 6 affected members, to be fundus flavimaculatus [35]. These individuals had initial good acuity, field and ERG, but in mid-life developed macular disease, with or without flecks or fundus flavimaculatus, with reduction of acuity, central/pericentral scotomas, and a modestly reduced ERG. In late life more generalized pigment epithelial loss with damage to overlying photoreceptors occurs with greater loss of acuity, field abnormalities, and ERG loss, often with marked regional or diffuse loss of choriocapillaris. Although Lam et al. used the term dominant retinitis pigmentosa to describe their case, possibly because of some bone-spicules pigmentation in the mid-peripheral fundus, the proband was asymptomatic until age 44 years, the visual acuity was 20/20, the ERG only modestly abnormal, and the decrease in peripheral field mild [53].
Jacobson et al. reported detailed studies of photoreceptor function in heterozygotes with three presumed null alleles including 1-bp ins, codon 32, FS44ter, 2-bp del, codon 193, FS216ter, and 5-bp del, 8-bp ins, codon 67-69, producing Met67Arg, Gly68His, and an Arg insertion at position 69 [57]. All showed more pericentral than peripheral field loss, normal ERG a-waves in younger subjects, yellow macular fleck-like deposits in mid-life, and equal cone and rod ERG loss in older individuals.
Pattern macular dystrophy has been associated with a 4 bp insertion at codon 140 that 8 amino acids later leads to a premature termination codon [49,50]. Butterfly-shaped pigment dystrophy of the fovea has been reported with a 2 bp deletion beginning with the third bp of codon 299, creating a frame shift and a premature termination at codon 355 [46].
Conclusions on the Mechanism of Disease in Peripherin/RDS Mutations
Peripherin/rds appears essential for morphogenesis and stability of rod and cone outer segment disks. RDS mutations cause several phenotypes including peripheral and macular degenerations. Missense mutations within the D2 loop of RDS that are associated with dominant retinitis pigmentosa occur at amino acid residues (Leu126Arg, Leu185Pro, Pro210, Phe211, Ser212, Cys214, Pro216, Pro219) that are identical in rom-1, whereas at least two of the missense mutations that are associated with macular dystrophy occur at residues (Arg142 and Arg172) that are not conserved in rom-1. Interactions between mutant rds and normal rds, rom-1, or other proteins may lead to the early death of photoreceptors through a dominant-negative effect or the triggering of apoptosis, producing the phenotypes of RP and cone or cone-rod dystrophy. These phenotypes are characterized by early abnormality of the ERG.
Other more tolerated missense mutations and null mutations (the latter FROM insufficient gene product or haploinsufficiency) may result in shortened or distorted photoreceptor outer segments which are shed at a higher than normal rate Such abnormal turnover could lead to excessive lipofuscin LOAD on the retinal pigment epithelium and resulting macular/pattern dystrophy (including Stargardt and fundus flavimaculatus-like pictures), with an initially normal ERG but late equal loss of both the rod and cone ERG | | | Acknowledgements | | Supported in part by The Foundation Fighting Blindness and Research to Prevent Blindness. | | | References | 1. Molday, R.S., Hicks, D., Molday, L. Peripherin: a rim-specific membrane protein of rod outer segment discs. Invest Ophthalmol Vis Sci 28:50-61 (1987).
2. Travis, G.H., Sutcliffe, J.G., Bok, D. The retinal degeneration slow (rds) gene product is a photorecptor disc membrane-associated glycoprotein. Neuron 6:61-70 (1991).
3. Arikawa, K., Molday, L.L., Molday, R.S., Williams, D.S. Localization of peripherin/rds in the disk membranes of cone and rod photoreceptors: relationship to disk membrane morphogenesis and retinal degeneration. J Cell Biol 116:659-667 (1992).
4. Travis, G.H., Christerson, L., Danielson, P.E., Klisak, I., Sparkes, R.S., Hahn, L.B., Dryja, T.P., Sutcliffe, J.G. The human retinal degeneration slow (RDS) gene: chromosome assignment and structure of the mRNA. Genomics 10:733-739 (1991).
5. Connell, G.J., Molday, R.S. Molecular cloning, primary structure, and orientation of the vertebrate photoreceptor cell protein peripherin in the rod outer segment disk membrane. Biochemistry 29:4691-4698 (1990).
6. Bascom, R.A., Manara, S., Collins, L., Molday, R.S., Kalnins, V.I., McInnes, R.R. Cloning of the cDNA for a novel photoreceptor membrane protein (rom-1) identifies a disk rim protein family implicated in human retinopathies. Neuron 8:1171-1184 (1992).
7. Goldberg, A.F.X., Moritz, O.L., Molday, R.S. Heterologous expression of photoreceptor peripherin/rds and rom-1 in COS-1 cells: Assembly, interactions and localization of multisubunit complexes. Biochemistry 34:14213-14219 (1995).
8. Kedzierski, W., Moghrabi, W.N., Allen, A.C., Jablonski-Stiemke, M., Bok, D., Travis, G.H. Three homologs of rds/peripherin in Xenopus laevis photoreceptors that exhibit covalent and non-covalent interactions. J. Cell Sci 109:2551-2560 (1996).
9. Moritz, O.L., Molday, R.S. Molecular cloning, membrane topology, and localization of bovine rom-1 in rod and cone photoreceptor cells. Invest Ophthalmol Vis Sci 37:352-362 (1996).
10. Chen, J., Flannery, J.G., LaVail, M.M., Steinberg, R.H., Xu, J., Simon, M.I. bcl-2 overexpression reduces apoptotic photoreceptor cell death in three different retinal degenerations. Proc Natl Acad Sci USA 93:7042-7047 (1996).
11. Travis, G.H., Brennan, M.B., Danielson, P.E., Kozak, C.A., Sutcliffe, J.G. Identification of a photoreceptor-specific mRNA encoded by the gene responsible for retinal degeneration slow (rds). Nature 338:70-73 (1989).
12. Molday, R.S. Peripherin/rds and rom-1: Molecular properties and role in photoreceptor cell degeneration. In: Chader G, Osborne N, ed. Progress in Retinal and Eye Research. London: Pergamon Press, vol 13, pp. 271-299 (1994).
13. Ma, J., Norton, J.C., Allen, A.C., Burns, J.B., Hasel, K.W., Burns, J.L., Sutcliffe, J.G., Travis, G.H. Retinal degeneration slow (rds) in mouse results FROM simple insertion of a t haplotype-specific element INTO protein-coding exon II. Genomics 28:212-219 (1995).
14. Jansen, H.G., Sanyal, S. Development and degeneration of retina in rds mutant mice: electron microscopy. J Comp Neurol 224:71-84 (1984).
15. Hawkins, R.K., Jansen, H.G., Sanyal, S. Development and degeneration of retina in rds mutant mice: photoreceptor abnormalities in the heterozygotes. Exp Eye Res 41:701-720 (1985).
16. Kedzierski, W., Lloyd, M., Birch, D.G., Bok, D., Travis, G.H. Generation and analysis of transgenic mice expressing P216L-substituted rds/peripherin in rod photoreceptors. Invest Ophthalmol Vis Sci 38:498-509 (1997).
17. Steinberg, R.H., Fisher, S.K., Anderson, D.H. Disc morphogenesis in vertebrate photoreceptors. J Comparative Neurology 190:501-518 (1980).
18. Corless, J.M., Fetter, R.D., Costello, M.J. Structural features of the terminal loop region of frog retinal rod outer segment disk membranes: I. Organization of lipid components. J Comp Neurol 257:1-8 (1987).
19. Corless, J.M., Fetter, R.D., Zampighi, O.B., Costello, M.J., Wall-Buford, D.L. Structural features of the terminal loop region of frog retinal rod outer segment disk membranes: II. Organization of the terminal loop complex. J Comp Neurol 257:9-23 (1987).
20. Corless, J.M., Fetter, R.D. Structural features of the terminal loop region of frog retinal rod outer segment disk membranes: III. Implications of the terminal loop complex for disk morphogenesis, membrane fusion, and cell surface interactions. J Comp Neurol 257:24-38 (1987).
21. Kajiwara, K., Hahn, L.B., Mukai, S., Travis, G.H., Berson, E.L., Dryja, T.P. Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature 354:480-483 (1991).
22. Farrar, G.J., Jordan, S.A., Kenna, P., Humphries, M.M., Kumar-Singh, R., McWilliam, P., Allamand, V., Sharp, E., Humphries, P. Autosomal dominant retinitis pigmentosa: localisation of a disease gene (RP6) to the short arm of chromosome 6. Genomics 11:870-874 (1991).
23. Farrar, G.J., Kenna, P., Jordan, S.A., Kumar-Singh, R., Humphries, M.M., Sharp, E.M., Sheils, D.M., Humphries, P. A three-base-pair deletion in the peripherin-RDS gene in one form of retinitis pigmentosa. Nature 354:478-480 (1991).
24. Wells, J., Wroblewski, J., Keen, J., Inglehearn, C., Jubb, C., Eckstein, A., Jay, M., Arden, G., Bhattacharya, S., Fitzke, F., Bird, A. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nature Genetics 3:213-218 (1993).
25. Kajiwara, K., Berson, E.L., Dryja, T.P. Screen for mutations in the entire coding sequence of the human rds/peripherin gene in patients with hereditary retinal degenerations. Invest Ophthalmol Vis Sci 34(suppl):1149 (1993).
26. Kemp, C.M., Jacobson, S.G., Cideciyan, A.V., Kimura, A.E., Sheffield, V.C., Stone, E.M. RDS gene mutations causing retinitis pigmentosa or macular degeneration lead to the same abnormality in photoreceptor function. Invest Ophthalmol Vis Sci 35:3154-3162 (1994).
27. Gorin, M.B., Jackson, K.E., Ferrell, R.E., Sheffield, V.C., Jacobson, S.G., Gass, J.D.M., Mitchell, E., Stone, E.M. A peripherin/Retinal Degeneration Slow mutation (Pro-210-Arg) associated with macular and peripheral retinal degeneration. Ophthalmology 102:246-255 (1995).
28. Ekström, U., Ponjavic, V., Abrahamson, M., Nilsson-Ehle, P., Andréasson, S., Stenström, I., Ehinger, B. Phenotypic expression of autosomal dominant retinitis pigmentosa in a Swedish family expressing a Phe-211-Leu variant of peripherin/RDS. In: Ponjavic V, ed. Phenotypes and Genotypes in families with Hereditary Tapetoretinal Degenerations. Lund: Grahns Boktryckeri, pp. 135-153 (1997).
29. Farrar, G.J., Kenna, P., Jordan, S.A., Kumar-Singh, R., Humphries, M.M., Sharp, E.M., Sheils, D., Humphries, P. Autosomal dominant retinitis pigmentosa: A novel mutation at the peripherin/RDS locus in the original 6p-linked pedigree. Genomics 14:805-807 (1992).
30. Saga, M., Mashima, Y., Akeo, K., Oguchi, Y., Kudoh, J., Shimizu, N. A novel Cys-214-Ser mutation in the peripherin/RDS gene in a Japanese family with autosomal dominant retinitis pigmentosa. Hum Genet 92:519-521 (1993).
31. Fishman, G.A., Stone, E., Gilbert, L.D., Vandenburgh, K., Sheffield, V.C., Heckenlively, J.R. Clinical features of a previously undescribed codon 216 (proline to serine) mutation in the peripherin/retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Ophthalmology 101:1409-1421 (1994).
32. Kikawa, E., Nakazawa, M., Chida, Y., Shiono, T., Tamai, M. A novel mutation (Asn244Lys) in the peripherin/RDS gene causing autosomal dominant retinitis pigmentosa associated with bull's-eye maculopathy detected by nonradioisotopic SSCP. Genomics 20:137-139 (1994).
33. Nakazawa, M., Kikawa, E., Kamio, K., Chida, Y., Shiono, T., Tamai, M. Ocular findings in patients with autosomal dominant retinitis pigmentosa and transversion mutation in codon 244 (Asn244Lys) of the peripherin/RDS gene. Arch Ophthalmol 112:1567-1573 (1994).
34. Nakazawa, M., Kikawa, E., Chida, Y., Wada, Y., Shiono, T., Tamai, M. Autosomal dominant cone-rod dystrophy associated with mutations in codon 244 (Asn244His) and codon 184 (Tyr184Ser) of the peripherin/RDS gene. Arch Ophthalmol 114:72-78 (1996).
35. Apfelstedt-Sylla, E., Theischen, M., Rüther, K., Wedemann, H., Gal, A., Zrenner, E. Extensive intrafamilial and interfamilial phenotype variation among patients with autosomal dominant retinal dystrophy and mutations in the human RDS/peripherin gene. Br J Ophthalmol 79:28-34 (1995).
36. Kajiwara, K., Sandberg, M.A., Berson, E.L., Dryja, T.P. A null mutation in the human peripherin/RDS gene in a family with autosomal dominant retinitis punctata albescens. Nature Genetics 3:208-212 (1993).
37. Kajiwara, K., Berson, E.L., Dryja, T.P. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 264:1604-1608 (1994).
38. Clarke, G.A., Kedzierski, W., Rossant, J., Travis, G.H., McInnes, R.R. The genetics of ROM1: evaluation of the requirement for rom-1 in photoreceptor morphogenesis, and of the digenic hypothesis of retinitis pigmentosa. Am J Hum Genet 59 (suppl):A46 (1996).
39. Wroblewski, J.J., Wells, J.A., Eckstein, A., Fitzke, F., Jubb, C., Keen, T.J., Inglehearn, C., Bhattacharya, S., Arden, G.B., Jay, M., Bird, A.C. Macular dystrophy associated with mutations at codon 172 in the human retinal degeneration slow (rds) gene. Ophthalmology 101:12-22 (1994).
40. Nakazawa, M., Wada, Y., Tamai, M. Macular dystrophy associated with monogenic Arg172Trp mutation of the peripherin/RDS gene in a Japanese family. Retina 15:518-523 (1995).
41. Reig, C., Serra, A., Gean, E., Vidal, M., Arumí, J., De la Calzada, M.D., Antich, J., Carballo, M. A point mutation in the RDS-peripherin gene in a Spanish family with central areolar choroidal atrophy. Ophthalmic Genetics 16:39-44 (1995).
42. Piguet, B., Héon, E., Munier, F.L., Grounauer, P.A., Niemeyer, G., Butler, N., Schorderet, D.F., Sheffield, V.C., Stone, E.M. Full characterization of the maculopathy associated with an Arg-172-Trp mutation in the RDS/peripherin gene. Ophthalmic Genetics 17:175-186 (1996).
43. Downes, S.M., Payne, A.M., Bessant, D.A.R., Fitzke, F.W., Holder, G.E., Bhattacharya, S.S., Bird, A.C. The RDS 172 mutation shows a common phenotype in twelve different families. Invest Ophthalmol Vis Sci 38(4):S798 (1997).
44. Nichols, B.E., Kimura, A.E., Streb, L.M., Sheffield, V.C., Stone, E.M. Mutations in the rds gene are associated with butterfly-shaped pigment dystrophy of the fovea. Invest Ophthalmol Vis Sci 34(suppl):1149 (1993).
45. Nichols, B.E., Sheffield, V.C., Vandenburgh, K., Drack, A.V., Kimura, A.E., Stone, E.M. Butterfly-shaped pigment dystrophy of the fovea caused by a point mutation in codon 167 of the RDS gene. Nature Genetics 3:202-207 (1993).
46. Nichols, B.E., Drack, A.V., Vandenburgh, K., Kimura, A.E., Sheffield, V.C., Stone, E.M. A 2 base pair deletion in the RDS gene associated with butterfly-shaped pigment dystrophy of the fovea. Hum Mol Genet 2:601-603 (1993).
47. Fossarello, M., Bertini, C., Galantuomo, M.S., Cao, A., Serra, A., Pirastu, M. Deletion in the peripherin/RDS gene in two unrelated Sardinian families with autosomal dominant butterfly-shaped macular dystrophy. Arch Ophthalmol 114:448-456 (1996).
48. Feist, R.M., White, M.F., Jr, Skalka, H., Stone, E.M. Choroidal neovascularization in a patient with adult foveomacular dystrophy and a mutation in the retinal degeneration slow gene (Pro210Arg). Am J Ophthalmol 118:259-260 (1994).
49. Keen, T.J., Inglehearn, C.F., Kim, R., Bird, A.C., Bhattacharya, S. Retinal pattern dystrophy associated with a 4 bp insertion at codon 140 in the RDS-peripherin gene. Hum Mol Genet 3:367-368 (1994).
50. Kim, R.Y., Dollfus, H., Keen, T.J., Fitzke, F.W., Arden, G.B., Bhattacharya, S.S., Bird, A.C. Autosomal dominant pattern dystrophy of the retina associated with a 4 bp insertion at codon 140 in the RDS/peripherin gene. Arch Ophthalmol 113:451-455 (1995).
51. Weleber, R.G., Carr, R.E., Murphey, W.H., Sheffield, V.C., Stone, E.M. Phenotypic variation including retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of the peripherin/RDS gene. Arch Ophthalmol 111:1531-1542 (1993).
52. Meins, M., Grüning, G., Blankenagel, A., Krastel, H., Reck, B., Fuchs, S., Schwinger, E., Gal, A. Heterozygous "null allele" mutation in the human peripherin/RDS gene. Hum Mol Genet 2:2181-2182 (1993).
53. Lam, B.L., Vandenburgh, K., Sheffield, V.C., Stone, E.M. Retinitis pigmentosa associated with a dominant mutation in codon 46 of the peripherin/RDS gene (arginine-46-stop). Am J Ophthalmol 119:65-71 (1995).
54. Fishman, G.A., Stone, E.M., Alexander, K.R., Gilbert, L.D., Derlacki, D.J., Butler, N.S. Serine-27-phenylalanine mutation within the peripherin/RDS gene in a family with cone dystrophy. Ophthalmology 104:299-306 (1997).
55. Nakazawa, M., Naoi, N., Wada, Y., Nakazaki, S., Maruiwa, F., Sawada, A., Tamai, M. Autosomal dominant cone-rod dystrophy associated with a Val200Glu mutation of the peripherin/RDS gene. Retina 16:405-410 (1996).
56. Hoyng, C.B., Heutink, P., Testers, L., Pinckers, A., Deutman, A.F., Oostra, B.A. Autosomal dominant central areolar choroidal dystrophy caused by a mutation in codon 142 in the peripherin/RDS gene. Am J Ophthalmol 121:623-629 (1996).
57. Jacobson, S.G., Cideciyan, A.V., Kemp, C.M., Sheffield, V.C., Stone, E.M. Photoreceptor function in heterozygotes with insertion or deletion mutations in the RDS gene. Invest Ophthalmol Vis Sci 37:1662-1674 (1996).
58. Richards, S.C., Creel, D.J. Pattern dystrophy and retinitis pigmentosa caused by a peripherin/RDS mutation. Retina 15:68-72 (1995). | |
|
|
|
|
|
|
 Welcome, please sign in
Welcome, please sign in