|
|
 |
|
|
|
|
|
Digenic Inheritance of a ROM1 Gene Mutation With A Peripherin/RDS or Rhodopsin Mutation in Families with Retinis Pigmentosa
Digital Journal of Ophthalmology 1999
Volume 5, Number 6
December 10, 1999
|
Printer Friendly
|
Samuel G. Jacobson | University of Pennsylvania, Scheie Eye Institute Roderick R. McInnes | The Research Institute, Hospital for Sick Children Val C. Sheffield | University of Lund Berndt Ehinger | University of Lund Sten Andreasson | University of Lund Ulf Ekström | University of Lund Magnus Abrahamson | University of Lund Vesna Ponjavic | University of Lund Roger A. Bascom | The Research Institute, Hospital for Sick Children Artur V. Cideciyan | University of Pennsylvania, Scheie Eye Institute Edwin M. Stone | University of Iowa Hospitals and Clinics
|
|
|
| Abstract | Objective
Two families with retinitis pigmentosa showed inheritance of an Arg-16-His ROM1 gene mutation with either an Arg-13-Trp RDS mutation or an Arg-135-Trp RHO mutation. The phenotypes of double and single heterozygotes were determined to examine the hypothesis that digenic inheritance may increase disease expression. In the family with ROM1 and RDS mutations, single heterozygotes were normal but one double heterozygote showed severe RP. Two other double heterozygotes, however, were normal by clinical and retinal function tests. In the family with ROM1 and RHO mutations, single heterozygotes with the RHO mutation all manifested RP, while a single heterozygote for the ROM1 mutation was normal. Disease severity was comparable in double heterozygotes and single heterozygotes HAVING the RHO mutation. We conclude that the Arg-16-His ROM1 gene mutation is non-pathogenic in the single heterozygous state, and there is no consistent evidence of digenic augmentation of pathogenicity in double heterozygotes carrying the Arg-16-His ROM1 mutation with either the benign Arg-13-Trp RDS mutation or the disease-causing Arg-135-Trp RHO mutation.Keywords
retinitis pigmentosa, retina, histopathology | | | Introduction | The coincidence of certain peripherin/RDS (RDS) and ROM1 gene mutations in a limited number of families with retinitis pigmentosa (RP) has led to the hypothesis that these double heterozygous states can cause RP while heterozygotes with only the RDS or the ROM1 gene mutations are clinically unaffected.[1,2] This observation is significant because it could represent an example of polygenic, specifically digenic, causation in retinal degenerative disease.
In this study, we describe two families with RP in which there is coincidence of a ROM1 gene mutation and either an RDS or a rhodopsin (RHO) mutation. In one family, some members are double heterozygotes for a codon 16 ROM1 mutation and a codon 13 RDS gene mutation and other members are single heterozygotes for one or the other mutation. To test the hypothesis that digenic inheritance of these mutations can cause RP, we used clinical methods and visual function tests to determine the phenotype of the double heterozygotes and the single heterozygotes with these RDS or ROM1 gene mutations.
The same codon 16 ROM1 gene mutation was also found in a family with autosomal dominant RP that cosegregated with a codon 135 mutation in the RHO gene.[3] In this family, the presence of double and single heterozygotes for the ROM1 and RHO mutations provided an opportunity to inquire further about pathogenicity of the ROM1 mutation alone and to determine if there was any evidence of increased disease severity in the double heterozygotes compared to single heterozygotes for the RHO mutation only. | | | Materials and Methods | Two families were included in the study (Fig. 1). In Family 1, there were coincident ROM1 and RDS mutations. In Family 2,[3] there were coincident ROM1 and RHO mutations. The procedures used to screen for mutations in the RHO, RDS and ROM1 genes have been published.[3-6]
The patients had ocular examinations and visual function tests to determine the phenotype associated with the different genotypes. Goldmann kinetic perimetry was performed with V and I test targets at intensity 4e. Full field electroretinograms (ERGs) were elicited, recorded and analyzed using published methods.[7-9] The institutional review boards approved these studies and informed consent was obtained FROM all participants after full explanation of the studies were given. | |

Figure 1
Pedigrees of the two families in this study. Filled symbols are patients with retinal degeneration; unfilled symbols indicate no detectable disease. Notation for the genotypes is as follows: +/+, wild type for ROM1 and RDS in Family 1 and ROM1 and RHO in Family 2; ROM1/+, heterozygote for ROM1 mutation; RDS/+, heterozygote for RDS mutation and RHO/+, heterozygote for rhodopsin mutation.
|
|
| Results | Genotype
In Family 1, DNA sequence abnormalities in the RDS and ROM1 genes were detected. In the RDS gene, there was a cytosine to thymidine transition (CGG -> TGG) in codon 13. This results in an arginine to tryptophan change at this position in the protein (Arg13Trp). In the ROM1 gene a guanosine to adenosine transition was found (CGC -> CAC) in codon 16. This results in an arginine to histidine change at this position in the protein (Arg16His). These mutations were not found in 100 control alleles and in 1200 patients with retinal degeneration (in laboratories of Iowa City, Iowa, USA, and Toronto, Ontario, Canada). No mutations in the coding sequence of the RHO gene were found in this family.
In Family 2, DNA sequence abnormalities in the RHO and ROM1 genes were detected. A cytosine to thymidine transition (CGG -> TGG) in codon 135 of the RHO gene was found, resulting in an arginine to tryptophan change (Arg135Trp) as previously described.[3] Subsequent screening of the ROM1 gene led to the finding that some members of this family, like Family 1, also had the Arg16His mutation. Screening of certain exons of the RDS gene showed no mutations.[3]
Phenotype
In Family 1 (Fig.1), there were three double heterozygotes for RDS and ROM1 mutations (ages 21, 43 and 73 at most recent examinations), one single heterozygote for the RDS mutation (age 44), and a single heterozygote for the ROM1 mutation (age 17).
Patient I-1, the oldest double heterozygote, described slowly progressive loss of night vision and visual field. On an examination at age 50, she had 20/40 visual acuity and central islands of vision only; at age 73, visual acuities had decreased to light perception and visual fields were not measurable. There were pigmentary changes throughout her retina, including the macula, with attenuated vessels and waxy pallor of the optic nerve. Patches of chorioretinal atrophy were also present in the midperiphery of both eyes. Patient II-1, a double heterozygote, and her sibling II-2, who is heterozygous for the RDS mutation, had normal ocular examinations, normal visual acuity, and normal ERGs at the ages of 20 and 21 years respectively. A recent reevaluation, representing a 23 year follow-up, showed that both patients still had normal clinical examinations and visual function tests. Patient III-1, another double heterozygote, and his sibling III-2, a single heterozygote for the ROM1 gene mutation, also had normal examinations.
Kinetic visual fields and ERGs in the three double heterozygotes of Family 1 are shown in Figure 2. Both patients III-1 and II-1 have normal extent of visual field with both target sizes. All parameters of the ERGs are within normal limits for both patients (normal DATA published previously[7]). Patient I-1, on an examination at the age of 50 years, had a visual field with only some central islands of function and ERGs were not detectable to any stimuli.
In Family 2, there were three double heterozygotes for RHO and ROM1 mutations (ages 19, 31 and 50) and three single heterozygotes for the RHO mutation (ages 11, 16 and 40). Details of the disease manifestations in these six patients have been reported.[3] In general, these patients had a relatively severe form of RP with ERG evidence of no detectable rod function and reduced cone function. Patient IV-6, a single heterozygote for the ROM1 mutation had a normal ocular examination and normal visual function test results.
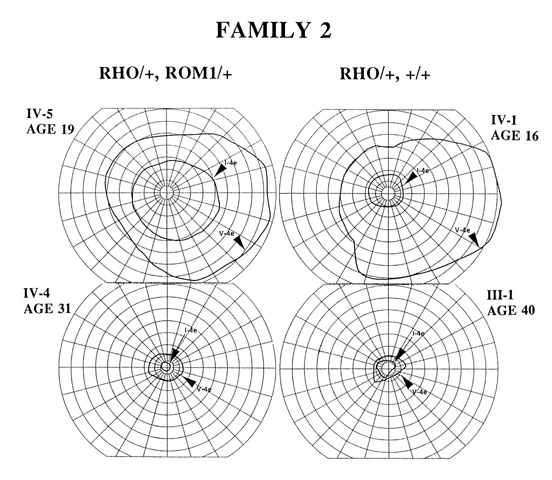
A comparison of kinetic fields in two double heterozygotes (RHO/+, ROM1/+) and two single heterozygotes (RHO/+, +/+) is shown in Figure 3. Patients IV-5 and IV-1 (ages 19 and 16, respectively), representing relatively early disease stages, do not differ dramatically enough in visual function to raise suspicion that the double heterozygote has greater disease expression than the single heterozygote. Another single heterozygote for the RHO mutation, IV-3 (age 11), had a comparable kinetic field (see Fig.3, ref.3). Patients IV-4 and III-1 (ages 31 and 40, respectively), at later stages of disease, do not SHOW remarkable differences in kinetic field extents | |

Figure 2
Kinetic visual fields (upper) and rod, mixed, and cone ERGs (lower) FROM the three double heterozygotes of Family 1. Arrows indicate stimulus onset. Calibrations are to the right and below the responses.
|
|

Figure 3
Comparison of kinetic visual fields for double (left) and single (right) heterozygotes in Family 2
|
|
| Discussion | Rom-1 and peripherin are membrane proteins located at the rim of the outer segments of photoreceptors. The ROM1 gene is on chromosome 11 and peripherin is encoded by the RDS (retinal degeneration slow) gene on chromosome 6.[10] Whereas heterozygous mutations in the RDS gene have been clearly associated with autosomal dominant retinal disease in man,[10-12] reports of definite associations between the ROM1 gene and retinopathy have not been forthcoming.[2,13-17]
The majority of RDS mutations involve the second intradiscal loop of the peripherin molecule with only a few in the transmembrane domains and at the carboxy terminal region. No disease-causing mutations have been reported to date at the amino terminal 20 amino acids.[10,12] The normal examination results in the heterozygote in Family 1 with the Arg13Trp RDS mutation, which would presumably affect the amino end of the molecule, suggests that this is not a pathogenic mutation in single dose. Based on the results FROM both families each with a single heterozygote with the Arg16His ROM1 mutation, this ROM1 gene change may also not be pathogenic. This latter conclusion is in keeping with recently published results of a small family with RP in which this missense change did not cosegregate with disease.[2]
The identification of these rare Arg13Trp RDS and Arg16His ROM1 alleles together in a patient with severe RP, however, leads to the notion that this may represent a form of "digenic RP", as reported in only four other families to date.[1,2] Unlike the previous reports, however, double heterozygosity (for the Arg13Trp RDS and the Arg16His ROM1 changes) was not sufficient to cause RP in all cases. Two further generations of double heterozygotes did not manifest disease.
What could be the reason for this difference between double heterozygosity in our patients and those with a Leu185Pro RDS and three different ROM1 gene mutations?[1,2] There is of course the possibility that the RDS and ROM1 gene mutations we detected in Family 1 are rare non-pathogenic sequence variants and have no relationship to the severe form of RP in patient I-1, which may be due to some other cause. The age of our patients could not be the reason. Our double heterozygotes with no manifest disease (21 and 43 years) were not younger than those with severe RP in the earlier report (age range, 18-52 years[1]). The two normal ocular examinations separated by 23 years in patient II-1 emphasizes that the natural history of this individual's condition is not that of a severe progressive retinal degeneration.
There is the possibility that different RDS and ROM1 mutations lead to different outcomes in terms of disease expression. The Leu185Pro RDS mutation and most of the associated ROM1 mutations[1,2] would be expected to involve the large second intradiscal loop between the third and fourth transmembrane segments of the molecules. This loop has been proposed to be the site of interactions between the two proteins at the disc rim.[5,10,18] Such mutations could interfere with photoreceptor structure and function more than the mutations identified in Family 1.
An alternative and more specific hypothesis, considering the rarity of reports to date of digenic RP, is that the phenomenon may be associated only with the Leu185Pro RDS mutation. This amino acid position in peripherin would thus be a key site of interest for study at the molecular level; to this purpose, the underlying pathogenic mechanisms have started to be explored.[19-21] A practical conclusion is that searching for RDS-ROM1 digenic causation in RP may not be worthwhile except when this codon 185 RDS mutation is detected in a family with RP. On the other hand, it should be recognized that the few families with convincing digenic causation have been found when sampling populations of patients with typical RP (defined mainly as severe disease).[1,2] Considering that many RDS-associated diseases are not in the category of typical RP,[12] and quantitation of the degree of photoreceptor dysfunction in these patients has been proven feasible,[7,22] it may be worthwhile to widen the search for digenic causation and use more than clinical criteria to define disease expression.
With only one affected member in Family 1, the statistically most likely diagnosis would be a form of autosomal recessive RP. However, if we speculate that patient I-1's form of RP is at least partly due to her RDS and ROM1 mutations, then an additional factor may also be required for the development of the retinal degeneration. An analogy can be drawn to the original digenic families,[1] in which a significant degree of incomplete penetrance was observed and later shown to be correlated with the absence of the ROM1 mutation. In Family 1 of this study, some double heterozygotes can also be considered to have incomplete penetrance and it seems reasonable to suspect the existence of a third genetic locus whose product interacts with or otherwise affects the pathogenicity of the RDS and ROM1 gene products. An interesting candidate gene to screen in this regard, for example, would be ABCR (ATP-binding cassette transporter) which encodes another photoreceptor-specific outer segment protein, incriminated in both inherited and age-related retinal degeneration.[23,24] The existence of a third locus is also supported by the finding of a family with RP of variable disease severity harboring one of the ROM1 mutations known to play a role in digenic RP but no detectable coding sequence mutation in the RDS gene.[15]
Rhodopsin, like rom-1 and peripherin, is a photoreceptor outer segment protein. The role of rhodopsin is not only structural but also functional; it is the light-absorbing visual pigment of the rods. Mutations in the rhodopsin gene are established causes of many forms of autosomal dominant RP and codon 135 mutations are among the more frequently reported worldwide.[25] Family 2 has a phenotype like that of other families described with the Arg135Trp mutation or other substitutions at this residue.[3] The finding of the coincident ROM1 mutation raised the theoretical possibility that the disease could have been accelerated by a potentially "digenic" effect. We found no evidence for such an effect in a small number of comparisons between single and double heterozygotes. The caveat is that an effect may have occurred but it may have eluded detection. If it did not augment the entire disease process but only the rod cell death, this could not have been observed in this particular mutation. Rod cell loss is a very early occurrence in patients with codon 135 mutations. At least, the 19 year old double heterozygote did not SHOW the same degree of degeneration as his older relatives.
In summary, this study found coincidence of mutations in the RDS and ROM1 genes and is the first to describe coincidence of RHO and ROM1 mutations. Our conclusion is that digenic enhancement of disease could not be consistently demonstrated in these families with these specific mutations. In the future when there are even greater numbers of genes known to cause or modify retinal degeneration and it becomes feasible to screen for all these genes simultaneously, the effects of multiply inherited gene changes on disease expression should be better understood. | | | Acknowledgements | | This collaborative study was stimulated by scientific interaction at the Erik K. Fernstrom Symposium on Tapetoretinal Degenerations (Lund, Sweden; June, 1997) and supported in part by U.S. Public Health Service Research Grants (EY05627, EY10539), The Foundation Fighting Blindness, The George Gund Foundation, The Chatlos Foundation, the RP Eye Research Foundation of Canada, the National Centres of Excellence Network, the Margit Thyselius fund, Sigvard and Marianne Bernadottes Forskningsstiftelse, the Faculty of Medicine at the University of Lund, the Swedish Society of Medicine and University Hospital of Lund. We are grateful to Dr. Jeremy Nathans for supplying RP patient DNA for this study; Ms. D. Suner, Ms. B. Koernig, and Ms. J. Lee for clinical coordination; and Mr. B. Eisner, Ms. K. Hummer, Mr. J. Christopher and Ms. K. Mejia for help with DATA analyses. | | | References | 1. Kajiwara, K., Berson, E.L., Dryja, T.P. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 264:1604-1608 (1994).
2. Dryja, T.P., Hahn, L.B., Kajiwara, K., Berson, E.L. Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 38:1972-1982 (1997).
3. Ponjavic, V., Abrahamson, M., Andreasson, S., Ehinger, B., Fex, G. Autosomal dominant retinitis pigmentosa with a rhodopsin mutation (Arg-135-Trp). Acta Ophthalmol. Scand. 75:218-223 (1997).
4. Nichols, B.E., Sheffield, V.C., Vandenburgh, K., Drack, A.V., Kimura, A.E., Stone, E.M. Butterfly-shaped pigment dystrophy of the fovea caused by a point mutation in codon 167 of the RDS gene. Nature Genet. 3:202-207 (1993).
5. Bascom, R.A., Schappert, K., McInnes, R.R. Cloning of the human and murine ROM1 genes: genomic organization and sequence conservation. Hum. Mol. Genet. 2:385-391 (1993).
6. Stone, E.M., Vandenburgh, K., Nichols, B.E., Sheffield, V.C. Identification of rhodopsin gene mutations using GC-clamped denaturing gradient gel electrophoresis. In: Hargrave P, ed. Photoreceptor Cells. Orlando, FL: Academic Press; 377-392 (1993).
7. Jacobson, S.G., Cideciyan, A.V., Kemp, C.M., Sheffield, V.C., Stone, E.M. Photoreceptor function in heterozygotes with insertion or deletion mutations in the RDS gene. Invest. Ophthalmol. Vis. Sci. 37:1662-1674 (1996).
8. Andreasson, S., Ehinger, B. Electroretinographic diagnosis in families with X-linked retinitis pigmentosa. Acta Ophthalmol. (Copenh) 68:139-144 (1990).
9. Andreasson, S., Ponjavic, V., Ehinger, B. Full field electroretinograms in a patient with cutaneous melanoma-associated retinopathy. Acta Ophthalmol. (Copenh) 71:487-490 (1993).
10. Molday, R.S. Peripherin/rds and rom-1: molecular properties and role in photoreceptor cell degeneration. In: Chader, G., Osborne, N., eds. Progress in Retinal and Eye Research. Great Britain: Pergamon Press Ltd; 271-299 (1994).
11. Bird, A.C. Retinal photoreceptor dystrophies. Am. J. Ophthalmol. 119:543-562 (1995).
12. Weleber, R.G. Phenotypic variation in patients with mutations in the peripherin/RDS gene. This volume (1997).
13. Bascom, R.A., Manara, S., Collins, L., Molday, R.S., Kalnins, V.I., McInnes, R.R. Cloning of the cDNA for a novel photoreceptor membrane protein (rom-1) identifies a disk rim protein family implicated in human retinopathies. Neuron 8:1171-1184 (1992).
14. Bascom, R.A., Liu, L., Humphries, P., Fishman, G.A., Murray, J.C., McInnes, R.R. Polymorphisms and rare sequence variants at the ROM1 locus. Hum. Mol. Genet. 2:1975-1977 (1993).
15. Sakuma, H., Inana, G., Murakami, A., Yajima, T., Weleber, R.G., Murphey W.H., Gass, J.D.M., Hotta, Y., Hayakawa, M., Fujiki, K., Gao, Y.Q., Danciger, M., Farber, D.B., Cideciyan, A.V., Jacobson, S.G. A heterozygous putative null mutation in ROM1 without a mutation in peripherin/RDS in a family with retinitis pigmentosa. Genomics 27:384-386 (1995).
16. Bascom, R.A., Liu, L., Heckenlively, J.R., Stone, E.M., McInnes, R.R. Mutation analysis of the ROM1 gene in retinitis pigmentosa. Hum. Molec. Genet. 4:1895-1902 (1995).
17. Martinez-Mir, A., Vilela, C., Bayes, M., Valverde, D., Dain, L., Beneyto, M., Marco, M., Baiget, M., Grinberg, D., Balcells, S., Gonzalez-Duarte, L.V. Putative association of a mutant ROM1 allele with retinitis pigmentosa. Hum. Genet. 99:827-830 (1997).
18. Molday, R.S. Molecular properties of the peripherin/rds-rom-1 complex and its role in retinal degenerative diseases. Great Basin Visual Science Symposium 2:52-61 (1996).
19. Goldberg, A.F.X., Molday, R.S. Defective subunit assembly underlies a digenic form of retinitis pigmentosa linked to mutations in peripherin/rds and rom-1. Proc. Natl. Acad. Sci. USA 93:13726-13730 (1996).
20. Clarke, G.A., Kedzierski W., Rossant, J., Travis, G.H., McInnes, R.R. The genetics of ROM1: evaluation of the requirement for rom-1 in photoreceptor morphogenesis, and of the digenic hypothesis of retinitis pigmentosa (RP). Am. J. Hum. Genet. 59 (suppl):A46 (1996).
21. Birch, D.G., Locke, K., Clarke, G., McInnes, R.R., Travis, G.H. ERGs in mice with RDS/peripherin and ROM1 mutations. Invest. Ophthalmol. Vis. Sci. 38:S316 (1997).
22. Jacobson, S.G., Cideciyan, A.V., Maguire, A.M., Bennett, J., Sheffield, V.C., Stone, E.M. Preferential rod and cone photoreceptor abnormalities in heterozygotes with point mutations in the RDS gene. Exp. Eye Research 63:603-608 (1996).
23. Allikmets, R., Singh, N., Sun, H., Shroyer, N.F., Hutchinson, A., Chidambararr, A., Gerrard, B., Baird, L., Stauffer, D., Peiffer, A., Rattner, A., Smallwood, P., Li, Y., Anderson, K.L., Lewis, R.A., Nathans, J., Leppert, M., Dean, M., Lupski, J.R. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nature Genet. 15:236-246.
24. Allikmets, R., Shroyer, N.F., Singh, N., Seddon, J.M., Lewis, R.A. Bernstein, P.S., Peiffer, A., Zabriski, N.A., Li, Y., Hutchinson, A., Dean, M., Lupski, J.R., Leppert, M. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 277:1805-1807 (1997).
25. Gal, A., Apfelstedt-Sylla, E., Janecke, A.R., Zrenner, E. Rhodopsin mutations in inherited retinal dystrophies and dysfunctions. Progress in Retinal and Eye Research 16:51-79 (1997). | |
|
|
|
|
|
|
 Welcome, please sign in
Welcome, please sign in