|
|
 |
|
|
|
|
|
Histopathology of Human Tapetoretinal Degenerations
Digital Journal of Ophthalmology 1998
Volume 4, Number 12
December 30, 1998
|
Printer Friendly
|
Amy H. Milam | University of Pennsylvania, Scheie Eye Institute, Philadelphia, PA, USA Zong-Yi Li | University of Washington, Seattle WA, USA Robert N. Fariss | University of Washington, Seattle WA, USA
|
|
|
| Abstract | Objective
Retinitis pigmentosa (RP) is a heterogeneous GROUP of inherited diseases that cause degeneration of photoreceptors in the human retina. Although the list of gene defects associated with RP is rapidly expanding, it is not known how any of the mutations leads to slowly progressive dysfunction and finally, death of the rods and cones. To provide this information, the retinas of patient donors with different genetic forms of RP have been evaluated by light and electron microscopy. Striking alterations have been found in mutant rods in the RP retinas, including outer segment shortening and sprouting of prominent, axon-like neurites. The rod neurites project for considerable distances INTO the inner layers of the RP retinas and are closely associated with hypertrophied Müller cell processes and GABA-positive amacrine cells. Loss of photoreceptor cells in the RP retinas, as well as secondary alterations in the retinal pigment epithelium, inner retinal neurons, glia and blood vessels are important considerations for understanding and developing therapies for this disease. | | | Introduction | The term retinitis pigmentosa (RP) encompasses a heterogeneous GROUP of inherited diseases that cause degeneration of rods and secondary but critical degeneration of cones in the human retina. Initial symptoms of RP, often noted in adolescence, are night vision problems and midperipheral visual field loss. The disease is progressive, and by adulthood, the more peripheral visual fields and macular function are usually reduced as well. In advanced RP, the retinal vessels are attenuated, black bone spicule pigment is deposited in the retina, and the optic nerve head appears pale and waxy.
The initial night vision problems are caused by dysfunction of the rods. Visual field loss is caused by death of the rods and cones, which for unknown reasons often begins in the equatorial region. Later death of macular photoreceptors reduces the central vision, although some RP patients retain relatively good visual acuity until late in life. Loss of rods and cones is accompanied by alterations in the RPE cells and retinal glia, and ultimately, in the inner retinal neurons and blood vessels.
Current strategies for RP therapy include transplantation of normal photoreceptors and RPE cells [1, 2], intravitreal injection of factors that prolong photoreceptor survival [3], delivery of corrective genes to mutant photoreceptors and RPE cells by means of viral vectors [4, 5] and liposomes [6], electrical stimulation of surviving neurons in the inner retina [7] and oral administration of vitamin A [8]. This paper is an abbreviated version of a longer review [9] of the histopathologic changes in human RP retinas. | |
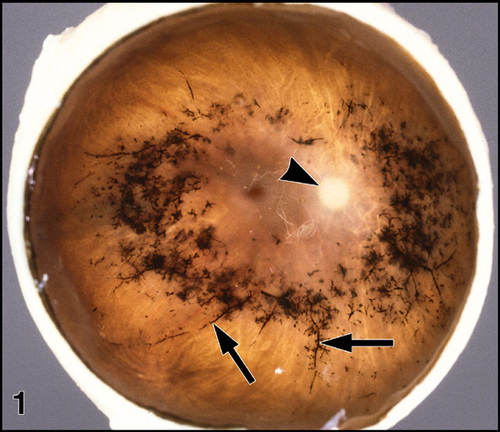
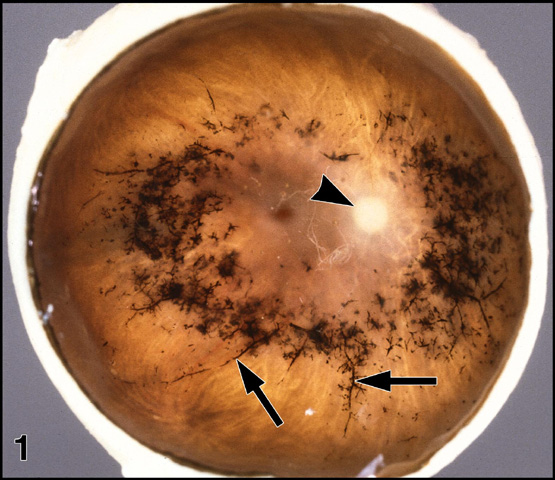
Figure 1
Gross pathology of an eye with RP (FFB#475). The anterior segment has been removed and the retina is viewed en face. Note deposits (arrows) of black bone spicule pigment around the retinal blood vessels. The arrowhead indicates the pale optic nerve head. Enlargement (116kbyte). Figure FROM [9] with permission of Pergamon Press.
|
|
| Results | Initial degenerative changes in RP retinas occur in the rods, followed by secondary changes in the cones, RPE, inner retinal neurons and glia, and retinal blood vessels. These retinal changes secondary to death of the rods are important considerations for therapies aimed at promoting the survival of mutant photoreceptors, replacing them with normal photoreceptors, or simulating their function by direct electrical stimulation of surviving retinal ganglion cells.
Photoreceptor transplantation procedures may be complicated by the patient's immune response to the introduced foreign cells, and this process may be exacerbated if inner retinal vessels are leaky in response to migrated RPE cells. Other features of the retinal milieu may be altered by the RP disease process such that transplanted rods either fail to survive or respond abnormally, for example by sprouting neurites and failing to make functional synapses with the correct INL neurons. Transplantation of normal photoreceptors INTO retinal regions that have formed bone spicule pigment will be complicated by the relocation of the host RPE cells to the inner retina, loss of the choriocapillaris, scar tissue formation by the Müller cells, and degenerative changes in the inner retinal blood vessels and neurons.
Insertion of normal genes INTO mutant rods or transplantation of normal rods to provide putative survival factors for surviving cones may be less affected by these retinal changes, although the surgical procedures will be complicated by shrinkage of the subretinal space as photoreceptor outer segments shorten. A major problem with the strategy to stimulate ganglion cells is the progressive loss of these cells caused by transneuronal degeneration and reduction in their blood supply. However, it was suggested [7] that preservation of even a small percentage of the retinal ganglion cells might be adequate to restore useful vision, based on restoration of auditory function to some deaf patients by stimulation of their partially degenerate auditory nerves with cochlear implants.
Other therapies on the horizon may be less affected by the secondary disease processes in the RP retinas. These include intravitreal injection of survival factors [3] and oral administration of vitamin A [8]. The mechanism of photoreceptor death in human RP [40], as in several animal models of retinal dystrophy, is thought to involve apoptosis or programmed cell death. Pharmacologic agents being developed to block apoptosis in other cells, for example central nervous system neurons, may also prolong photoreceptor life in RP retinas. Success of these experimental therapies will depend on increased understanding of the causes and mechanisms of death in mutant photoreceptors, as well as elucidation of the characteristic secondary changes in human RP retinas. We expect that continued identification of specific gene mutations in the RP patient donors will increase our understanding of the basis of retinal degeneration, information essential for developing effective therapies for this important GROUP of blinding diseases. | | | Discussion | Rod Photoreceptors
Histopathologic studies have been performed on retinal specimens FROM patient donors with different genetic forms of RP, including autosomal dominant RP due to mutations in the gene for the rod visual pigment, rhodopsin: proline-23-histidine [10], arginine-135-tryptophan [11], threonine-17-methionine [12], and glutamine-64-ter [13]. The process of rod cell death usually begins in the midperipheral retina and progresses with time to involve the macula and more peripheral retina, often sparing rods in the far periphery until late in the disease. These regional differences suggest that factor(s) other than the RP gene defect may influence the rate of photoreceptor cell death. Sparing of rods in the far periphery may correlate with lower levels of incident light [12] and the presence in this region [14] of the highest percentage of rods immunoreactive for basic fibroblast growth factor (bFGF), a peptide known to promote rod cell survival in dystrophic rodent retinas [3].
Outer Segments
In all genetic forms of RP, the earliest histopathologic change in the rods is outer segment shortening, visualized by immunocytochemistry using anti-rhodopsin (Figure 2).
In normal rods, rhodopsin is synthesized in the rough endoplasmic reticulum, transported in Golgi-derived vesicles through the inner segment to the connecting cilium, and inserted INTO newly forming membranes at the base of the outer segment [15]. The synthesis of large amounts of rhodopsin and other outer segment proteins is balanced by periodic shedding of outer segment tips, which are phagocytosed and degraded by the RPE [16]. Because large amounts of these outer segment proteins must be metabolized, mutant molecules that fail to undergo normal cellular processing may accumulate abnormally, leading to rod cell death.
In the case of mutant forms of rhodopsin, the abnormal molecules may fail to reach the outer segment because they are not transported out of the endoplasmic reticulum or Golgi apparatus, or are recognized a foreign molecules and degraded by lysosomal activity in the inner segment [17-19]. Mutations affecting amino acids near the N-terminus may interfere with glycosylation and incorporation of rhodopsin INTO forming outer segment discs [20]. However, accumulations of rhodopsin the rod inner segments have not been found in the rhodopsin-mutant human retinas studied to date [12, 13]. Rhodopsin-positive inclusions have been noted in rod inner segments of young transgenic pigs expressing proline-347-leucine rhodopsin [21], suggesting that only certain of the mutant forms of rhodopsin undergo abnormal accumulation.
Rod Neurites
Rods in RP retinas sprout long, axon-like neurite processes [13, 22] that contain rhodopsin and project for considerable distances INTO the inner retina (Figure 2). The rod neurites have been documented in retinas with RP caused by rhodopsin mutations and other genetic forms of RP, including XL RP where the rhodopsin gene is presumably normal [22]. Rod neurite sprouting is common in the peripheral regions of RP retinas where some death of photoreceptors has already occurred, but rod neurites have not been found in the maculas of the same retinas, even when this region has undergone significant photoreceptor loss. Cones do not undergo robust neurite sprouting, although some peripheral cone axons are elongated and appear to reach the inner plexiform layer (IPL) [13, 22].
The rod neurites bypass the dendrites of horizontal and rod bipolar cells (Figure 2), the normal targets of rod axons in the outer plexiform layer, and are closely associated with the hypertrophied processes of Müller cells that have undergone gliosis in response to photoreceptor death (Figure 3).
Electron microscopy reveals that the rod neurites contain multivesicular bodies, along with numerous small vesicles as found in normal rod synapses, but they do not appear to form true synapses with inner retinal neurons [22].
Rods neurite sprouting in vivo suggests that mature photoreceptors retain remarkable plasticity. Rods form neurites in vitro when cultured on Müller glia or NCAM [23-26], and the close association between rod neurites and hypertrophied Müller processes in the RP retinas suggests that neurite sprouting may be a response to novel Müller surface molecules expressed during the process of reactive gliosis. Neurite sprouting may also be a response to increased expression of one or more growth factors during photoreceptor degeneration, as occurs in dystrophic rodent retinas [27]. Other, as yet unknown alterations in RP retinas may also stimulate rods to form these remarkable neurite processes.
In dystrophic rds mouse retinas, rod synapses expand in size in response to loss of neighboring synapses [28]. However, rod neurite sprouting has not been documented in the rodent models for RP, where rod cell death is usually complete within days to months, as opposed to years to decades in human RP retinas [22]. Rod neurites have been found recently in the dystrophic retinas of longer lived animals, e.g., cats [29] and pigs [21]. Based on current information, rod neurite sprouting appears to be peculiar to mutant rods or represent a response to changes in the microenvironment unique to the degenerating retinas.
Such changes in the retinal milieu may have consequences for RP therapy involving photoreceptor transplantation [30]. One goal of this approach is to establish synapses between transplanted normal photoreceptors and the hostís surviving inner retinal neurons, recreating a functional visual pathway. Changes in the human RP retinal microenvironment that elicit robust neurite sprouting by the host mutant rods may have similar effects on transplanted photoreceptors. For example, a putative increase in growth factor(s) in the diseased human RP retinas may promote survival of the introduced photoreceptors but also lead to rod neurite sprouting and cone axonal abnormalities as found in RP retinas [13, 22]. Müller cell changes secondary to photoreceptor death may promote similar changes in transplanted photoreceptors. Because the rod neurites in the RP retinas project past the neurons that normally receive synaptic input FROM rod axons [22], normal rods transplanted INTO such retinas may not receive signals for axon termination and synapse formation required for their functional integration. Clearly, more complete characterization of the RP retinas is needed as a foundation for developing this form of therapy.
Cone Photoreceptors
Rod cell death in RP retinas is usually accompanied by changes in neighboring cones, including outer segment shortening, cytoplasmic densification, axonal elongation, and ultimately, cone cell death [12, 13]. Cone degeneration occurs in all genetic forms of RP, including AD RP caused by mutations in a rod-specific gene, e.g., rhodopsin, which is presumably not expressed in the cones. It was suggested that cone cell death may be triggered by toxic byproducts of rod cell degeneration [18] or result FROM loss of trophic factors for cones derived normally FROM rods [2, 31].
An important aspect of the RP disease process is loss of macular function. While loss of rod function constitutes an inconvenience for RP patients, loss of cone-mediated macular function significantly decreases their quality of life. Most RP maculas SHOW loss of both rods and cones, often retaining only a monolayer of cone somata with very short or absent outer segments [13, 32]. Peripheral cones also SHOW outer segment shortening in areas where rods have begun to degenerate.
RPE, Bruchís Membrane, and Choriocapillaris
Following death of all photoreceptors, RPE cells often detach FROM Bruch's membrane and migrate to perivascular sites in the inner retina, producing bone spicule pigment [33]. This term refers to the black melanin granules in the RPE cells that surround branching retinal blood vessels, producing a spiculated appearance. The choriocapillaris is invariably missing FROM retinal regions that have lost photoreceptors and formed bone spicule pigment [33]. Because of the close interdependence of the choriocapillaris, RPE and photoreceptor cells, it is not clear if loss of this capillary bed is secondary to the death of the photoreceptors or absence of neighboring RPE cells; the latter appears likely because RPE loss in previously healthy animal eyes leads to secondary atrophy of the choriocapillaris [34]. The relocated RPE cells in the inner retina form epithelial cuffs around the blood vessels, most often the thin-walled capillaries and venules, which respond to the nearby RPE cells by developing endothelial fenestrations. The stimulus for RPE migration is unknown, but loss of metabolic photoreceptor byproducts may alter the adhesion of RPE cells to Bruch's membrane. The relocation of RPE cells to perivascular regions in the inner retina may reflect their affinity for vascular basal laminae [33].
This process of RPE migration is an obvious consideration for therapies involving transplantation, because RPE cells as well as photoreceptors must be replaced in the retinal regions that have formed bone spicule pigment. Choriocapillaris loss of the choriocapillaris in such regions is another important consideration. A recent study in pigs showed choriocapillaris atrophy within a week following RPE debridement [34]. It is unknown if the choriocapillaris can be reestablished after long standing loss of the RPE in areas of bone spicule pigment. Obviously this capillary bed is essential for survival of transplanted photoreceptors and RPE cells.
Müller Cells
Müller cells undergo reactive gliosis in a variety of retinal degenerations, including RP [12, 22, 35]. The gliotic changes in Müller cells include hypertrophy, migration of enlarged nuclei to the outer retina, and increased immunoreactivity for glial fibrillary acid protein (GFAP).
In advanced RP, the degenerated photoreceptors are replaced by a prominent layer of thickened Müller cell processes. Reactive gliosis by the Müller cells is an important consideration for therapy for RP retinas because a thick glial scar in the outer retina may constitute a mechanical barrier to synapse formation between transplanted photoreceptors and inner nuclear layer (INL) neurons. The intimate association between rod neurites and gliotic Müller cell processes suggests that photoreceptors transplanted INTO gliotic RP retinas may sprout neurites rather than forming synapses with INL neurons.
Inner Nuclear Layer
The INL was studied in the maculas of 21 retinas with different genetic forms of RP. In spite of significant loss of macular photoreceptors and ganglion cells, retinas with severe RP showed retention of ~78% of INL cells [36]. The DATA suggested that photoreceptor loss caused only minimal transneuronal degeneration in the INL, but interpretation of this result was complicated by the fact that in addition to multiple neuron types (horizontal, bipolar, amacrine cells), the INL also contains the somata and nuclei of the Müller glia. If Müller cells undergo reactive hyperplasia in RP, as occurs, for example, in experimental retinal detachment [37], increased numbers of Müller nuclei in the INL might counterbalance a putative loss of INL neurons by transneuronal degeneration. To answer this question, additional morphometric studies are needed on RP retinas using specific markers for INL neurons and reactive Müller cells.
Preliminary evidence has been obtained [38] that like rods, certain inner neurons in RP retinas SHOW remarkable plasticity. GABA-positive amacrine cells sprout new processes that project well beyond their normal limits in the IPL. The GABA-positive amacrine somata also appear to be contacted preferentially by rhodopsin-positive rod neurites. Horizontal cells also sprout neurites that extend beyond the outer plexiform layer, coursing between remaining photoreceptors as far as the external limiting membrane. Secondary changes in the inner retina, including neuronal cell death and formation of novel neuronal processes, require further elucidation.
Retinal Ganglion Cells
Because RP leads to characteristic atrophy of retinal blood vessels and pallor of the optic nerve head, it has been the clinical impression that retinal ganglion cells are lost secondary to photoreceptor death [39]. Morphometric analysis of the maculas of 41 RP retinas [39] revealed significant reduction in ganglion cell numbers. In general, the loss of ganglion cells paralleled the loss of photoreceptors, and was more severe in XL and AD RP than in simplex RP [39].
A more recently recognized cause of ganglion cell death is relocation of RPE cells to perivascular sites in the inner retina. Some death of the inner neurons in the RP retinas is probably due to a decrease in their blood supply caused by RPE-induced alterations in the retinal vessels [33]. Such degenerative changes in the inner retinal neurons and blood vessels are important considerations for therapies based on photoreceptor transplantation or rescue, as well as electrical stimulation of surviving ganglion cells.
Retinal Blood Vessels
Attenuated retinal blood vessels, a funduscopic hallmark of RP, were historically thought to reflect diminished ganglion cell metabolism after photoreceptor death. We found migrated RPE cells in the inner retina produce remarkable alterations in the retinal blood vessels, including development of fenestrations in the endothelial cells [33]. Prominent extracellular matrix (ECM) deposits between the RPE and endothelial cells of the vessels closely resemble Bruch's membrane in situ, including two layers each of basal lamina and collagen, plus a middle layer of elastin not normally present in retinal vessels [33]. The perivascular ECM is often thick and contains prominent lipid and calcium deposits reminiscent of normal aging changes in Bruch's membrane. In extreme cases, these ECM deposits completely occlude the vessel lumina. Thickening of the walls of the blood vessels and occlusion of their lumina correlates with the retinal vessel sclerosis and atrophy that are characteristic of RP. The thick perivascular deposits, particularly those rich in hydrophobic lipid and elastin, may compromise the flow of nutrients FROM the vessel lumina to neurons in the inner retina. These degenerative changes in retinal blood vessels and inner neurons are important consideration for therapies based on the assumption that the inner layers of RP retinas are unaffected by death of the photoreceptors. | |
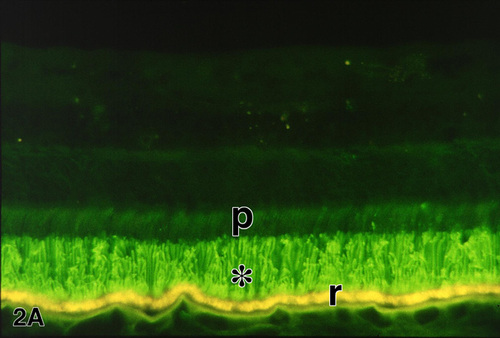
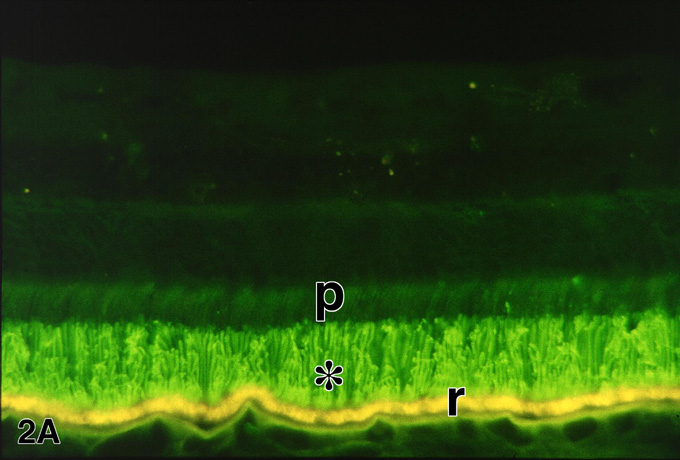
Figure 2a
Figures 2-2b. Immunofluorescence demonstration of rhodopsin in a normal retina (A) and the retina of an RP patient (FFB#424) (B) with autosomal dominant RP due to the glutamine-64-ter rhodopsin mutation. In the normal retina (A), rhodopsin immunolabeling (green) is restricted to the long rod outer segments (*); p, photoreceptor inner segments; r, RPE cells, which contain yellow-gold, autofluorescent lipofuscin granules. Enlargement of A (94 kbyte). In the RP retina (B), the rods are reduced to one or two rows of somata (arrowheads). Rhodopsin is delocalized to the surface membranes of the rod inner segments and somata, as well as long, beaded neurites (arrows) that extend INTO the inner retina. The rhodopsin-positive rod neurites bypass their normal targets in the inner nuclear layer, including the horizontal cells which are labeled (red) with anti-calbindin. Other calbindin-positive neurons in the inner nuclear layer are cone bipolar and amacrine cells. r, RPE. X 230. Enlargement of B (123 kbyte). Figure FROM [9] with permission of Pergamon Press.
|
|

Figure 2b
|
|
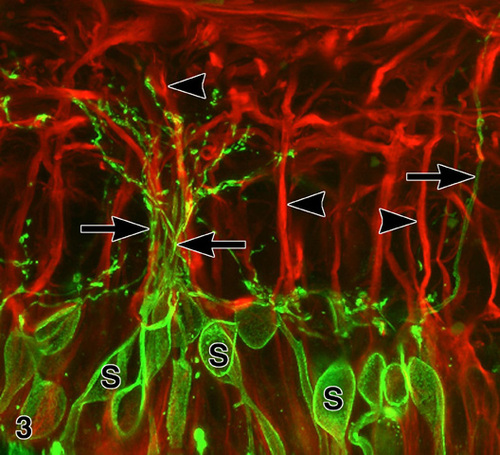
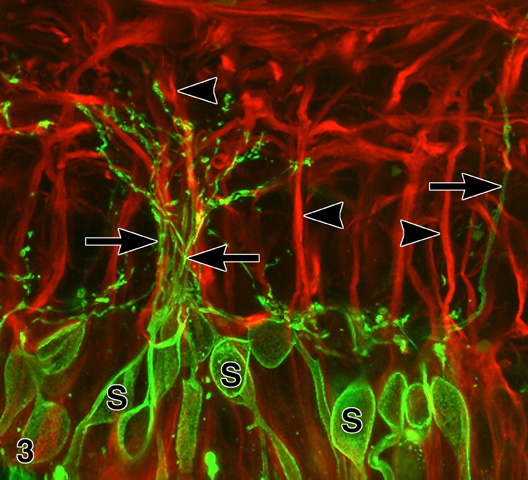
Figure 3
Confocal immunofluorescence microscopy of an RP retina (FFB#340) double labeled with anti-rhodopsin (green) and anti-GFAP (red). The rhodopsin-positive rod somata (s) give rise to neurites that run along the surfaces of GFAP(+) M¸ller cell processes (arrowheads). Note fascicle of rod neurites (between arrows). X 1,000. Enlargement (133 kbyte). Figure FROM [9] with permission of Pergamon Press.
|
|
| Acknowledgements | | This work was supported by The Foundation Fighting Blindness, Inc.; by NIH Grants EY0-1311 and -1730; by Research to Prevent Blindness, Inc.; and by the Chatlos Foundation. The authors thank Mr. D. Possin, Ms. I. Klock and Ms. J. Chang for technical assistance, and Mr. M. Stephens, Mr. R. Jones and B. Clifton for photographic help. | | | References | 1. Bok, D., Retinal Transplantation and Gene Therapy. Present Realities and Future Possibilities. Investigative Ophthalmology and Visual Science 34:473-476 [1993]
2. Milam, AH., Strategies for Rescue of Retinal Photoreceptor Cells. Current Opinion in Neurobiology 3:797-804. [1993]
3. Steinberg, RH., Survival Factors in Retinal Degenerations. Current Opinion in Neurobiology 4:515-524 [1994]
4. Bennett, J., Tanabe, T., Sun, D., Zeng, Y., Kjeldbye, H., Gouras, P., Maguire, AM., Photoreceptor Cell Rescue in Retinal Degeneration (rd) Mice by in vivo Gene Therapy. Nature Medicine 2:649-654 [1996]
5. Ali, RR., Reichel, MB., Thrasher, AJ., Levinsky, RJ., Kinnon, C., Kanuga, N., Hunt, DM., Bhattacharya, SS., Gene Transfer INTO the Mouse Retina Mediated by an Adeno-associated Viral Vector. Human Molecular Genetics 5:591-594 [1996]
6. Hangai, M., Kaneda, Y., Tanihara, H., Honda, Y., In vivo Gene Transfer INTO the Retina Mediated by a Novel Liposome System. Investigative Ophthalmology and Visual Science 37:2678-2685 [1996]
7. Humayun, M., de Juan Jr., E., Dagnelie, G., Greenberg, R., Propst, R., Phillips, H,. Visual Perception Elicited by Electrical Stimulation of Retina in Blind Humans. Archives of Ophthalmology 114:40-46 [1996]
8. Berson, EL., Rosner, B., Sandberg, MA., Hayes, KC., Nicholson, BW., Weigel-DiFranco, C., Willett, W., A Randomized Trial of Vitamin A and Vitamin E Supplementation for Retinitis Pigmentosa. Archives of Ophthalmology 111:761-772 [1993]
9. Milam, AH., Li, Z-Y., Fariss, RN., Histopathology of the Human Retina in Retinitis Pigmentosa. Progress in Retinal and Eye Research (In Press) [1997]
10. Kolb, H., Gouras, P., Electron Microscopic Observations of Human Retinitis Pigmentosa, Dominantly Inherited. Investigative Ophthalmology 13:487-498 [1974]
11. Tucker, GS., Jacobson, SG., Morphological Findings in Retinitis Pigmentosa with Early Diffuse Rod Dysfunction. Retina 8:30-41 [1988]
12. Li, Z-Y., Jacobson, SG., Milam, AH., Autosomal Dominant Retinitis Pigmentosa Caused by the Threonine-17-Methionine Rhodopsin Mutation: Retinal Histopathology and Immunocytochemistry. Experimental Eye Research 58:397-408 [1994]
13. Milam, AH., Li, Z-Y., Cideciyan, AV., Jacobson, SG., Clinicopathologic Effects of the Q64ter Rhodopsin Mutation in Retinitis Pigmentosa. Investigative Ophthalmology and Visual Science 37:753-765 [1996]
14. Li, Z-Y., Chang, JH., Milam, AH., A Gradient of Basic Fibroblast Growth Factor in Rod Photoreceptors in the Normal Human Retina. Visual Neuroscience (In Press) [1997]
15. Deretic, D., Papermaster, DS.. The Role of Small G-proteins in the Transport of Newly Synthesized Rhodopsin. In: Osborne, NN., Chader, GJ., eds. Progress in Retinal Research, Oxford, UK: Pergamon Press, 249-265. [1995]
16. Bok, D., Retinal Photoreceptor-Pigment Epithelium Interactions. Friedenwald Lecture. Investigative Ophthalmology and Visual Science 26:1659-1694. [1985]
17. Dryja, TP., Doyne Lecture. Rhodopsin and Autosomal Dominant Retinitis Pigmentosa. Eye 6:1-10 [1992]
18. Bird, AC., Investigation of Disease Mechanisms in Retinitis Pigmentosa. Ophthalmic Paediatrics and Genetics 13:57-66 [1992]
19. Manoil, C. Traxler, B., Membrane Protein Assembly: Genetic, Evolutionary and Medical Perspectives. Annual Review of Genetics 29:131-150 [1995]
20. Berson, EL., Ocular Findings in a Form of Retinitis Pigmentosa with a Rhodopsin Gene Defect. Transactions of the American Ophthalmological Society 88:355-388 [1990]
21. Li, Z-Y., Milam, AH., Chang, JH., Possin, DE., Hao, Y., Petters, RM., Wong, F., Characterization of Retinal Degeneration in Rhodopsin P347L Transgenic Pigs. Investigative Ophthalmology and Visual Science 38:S315 [1997]
22. Li, Z-Y., Kljavin, IJ., Milam, AH., Rod Photoreceptor Neurite Sprouting in Retinitis Pigmentosa. Journal of Neuroscience 15:5429-5438 [1995]
23. Kljavin, IJ., Reh, TA., Müller Cells are a Preferred Substrate for in vitro Neurite Extension by Rod Photoreceptor Cells. Journal of Neuroscience 11:2985-2994. [1991]
24. Mandell, JW., MacLeish, PR., Townes-Anderson, E., Process Outgrowth and Synaptic Varicosity Formation by Adult Photoreceptors in vitro. Journal of Neuroscience 13:3533-3548 [1993]
25. Kljavin, IJ., Lagenaur, C., Bixby, JL., Reh, TA., Cell Adhesion Molecules Regulating Neurite Growth FROM Amacrine and Rod Photoreceptor Cells. Journal of Neuroscience 14:5035-5049 [1994]
26. Hicks, D., Forster, V., Dreyfus, H., Sahel, J., Survival and Regeneration of Adult Human Photoreceptors in vitro. Brain Research 643:302-305 [1994]
27. Gao, H., Hollyfield, JG., Basic Fibroblast Growth Factor: Increased Gene Expression in Inherited and Light- induced Photoreceptor Degeneration. Experimental Eye Research 62:181-189 [1996]
28. Sanyal, S.. SynapticGrowth in the Rod Terminals after Partial PhotoreceptorLoss. In: Osborne, NN., Chader, GJ., eds. Progress in Retinal Research, Oxford, UK: Pergamon Press, 247-270. [1993]
29. Luthert, PJ., Chong, NHV., Barnett, KC., Bird, AC., An Immunohistochemical Study of the rdy Cat Model of Retinal Dystrophy. Investigative Ophthalmology and Visual Science 38:S311 [1997]
30. Bok, D., Hageman, GS., Steinberg, RH., REPAIR and Replacement to Restore Sight. Report FROM the Panel on Photoreceptor/Retinal Pigment Epithelium. Archives of Ophthalmology 111:463-471 [1993]
31. Huang, PC., Gaitan, AE., Hao, Y., Peters, RM., Wong, F., Cellular Interactions Implicated in the Mechanism of Photoreceptor Degeneration in Transgenic Mice Expressing a Mutant Rhodopsin Gene. Proceedings of the National Academy of Sciences, U.S.A. 90:8484-8488 [1993]
32. Milam, AH., Li, Z-Y.. Retinal Pathology in Retinitis Pigmentosa: Considerations for Therapy. In: Anderson, RE., Hollyfield, JG., LaVail, MM, eds. Retinal Degeneration II, New York, NY, USA: Plenum Press, 275-284 [1995]
33. Li, Z-Y., Possin, DE., Milam, AH., Histopathology of Bone Spicule Pigmentation in Retinitis Pigmentosa. Ophthalmology 102:805-816 [1995]
34. Del Priore, LV., Kaplan, HJ., Hornbeck, R., Jones, Z., Swinn, M., Retinal Pigment Epithelial Debridement as a Model for the Pathogenesis and Treatment of Macular Degeneration. American Journal of Ophthalmology 122:629-643 [1996]
35. Milam, AH., Jacobson, SG., Photoreceptor Rosettes with Blue Cone Opsin Immunoreactivity in Retinitis Pigmentosa. Ophthalmology 97:1620-1631 [1990]
36. Santos, A., Humayun, MS., de Juan, Jr., E., Greenberg, RJ., Marsh, MJ., Klock, IB., Milam, AH., Preservation of the Inner Retina in Retinitis Pigmentosa. A Morphometric Analysis. Archives of Ophthalmology 115:511-515 [1997]
37. Geller, SF., Lewis, GP., Anderson, DH., Fisher, SK., Use of the MIB-1 Antibody for Detecting Proliferating Cells in the Retina. Investigative Ophthalmology and Visual Science 36:737-744 [1995]
38. Fariss, RN., Li, Z-Y., Milam, AH., Evidence for Plasticity in Human RP Retinas. Investigative Ophthalmology and Visual Science 38:S260 [1997]
39. Stone, JL., Barlow, WE., Humayun, MS., de Juan Jr, E., Milam, AH., Morphometric Analysis of Macular Photoreceptors and Ganglion Cells in Retinas with Retinitis Pigmentosa. Archives of Ophthalmology 110:1634-1639 [1992]
40. Li, Z-Y., Milam, AH.. Apoptosis in Retinitis Pigmentosa. In: Anderson, RE., Hollyfield, JG., LaVail, MM., eds. Retinal Degeneration II, New York, NY, USA: Plenum Press, 1-12 [1995] | |
|
|
|
|
|
|
 Welcome, please sign in
Welcome, please sign in