Swaha Bose, MD, BMedSci | Ipswich General Hospital, Ipswich, Australia Damien C. M. Yeo, MBChB, FRCOphth | Paediatric Ophthalmology, Great Ormond Street Hospital for Children, London, United Kingdom Sidath Wijetilleka, MBBS, FRCOphth | Department of Ophthalmology, Singleton Hospital, Swansea, United Kingdom
Cystinosis, a rare autosomal recessive lysosomal storage disease, can be difficult to detect. The most common form of the disease is infantile or nephropathic cystinosis. Crystals can accumulate in the eye as early as 1 year of age. Early recognition and prompt investigations prevent further accumulation of cystine and resultant end-organ injury. The disease is usually confirmed through biochemical and genetic testing, which can be time consuming. Looking for cystine corneal deposits remains an important diagnostic criterion and is the least invasive test to perform. It is recommended that ophthalmic manifestations of cystinosis be confirmed by an ophthalmologist. We describe the case of a 3-year-old girl who presented with worsening emesis, pyrexia, and lethargy, and was diagnosed with infantile cystinosis. This case is used to present a technique that can facilitate the preliminary search for corneal cystine crystals by using equipment as readily available as two smartphones. The technique may be easily used in a variety of settings, including hospitals, clinics, and primary care centers where there is delayed or difficult access to ophthalmologists.
A 3-year-old girl presented with worsening emesis, pyrexia, and lethargy. Ten days prior, she had been diagnosed with a urinary tract infection after testing positive for urine erythrocytes, leukocytes, and protein at her local hospital. She was discharged on oral antibiotics. As her condition deteriorated, she was brought for evaluation to Great Ormond Street Hospital for Children. Serology revealed hyponatremia (116 mmol/L), hypokalemia (1.2 mmol/L), elevated C-reactive protein (25 mg/L), urea (20.8 mmol/L), and creatinine (210 µmol/L). She was diagnosed with renal tubular acidosis, exacerbated by infection, and admitted to the pediatric intensive care unit. Further questioning after admission revealed that she had poor oral intake and weight loss since the age of 12 months, and previous investigations in determining the cause of her failure to thrive were unsuccessful. On admission, her weight and height were in the 0.4th percentile.
Cystinosis was considered as a differential diagnosis for her renal tubulopathy. An ophthalmic consult was requested to seek evidence of pathognomonic corneal cystine crystals. In the first ward-based ophthalmology review, two smartphones were used to determine whether crystals were present. On examination, smartphone 1 was used as a mobile light-source and swiveled between 30° and 45° on its vertical axis to produce a specular reflection effect (Figure 1, Video 1). The device was held less than 10 cm from the eye and angled so that the light fell on the lateral periphery of the cornea (Figure 2). Smartphone 2 recorded a video close-up of the eye. This process captured “ground glass” corneal haziness in the affected eye.
As a result of corneal findings of this examination, cystinosis was further investigated. On ophthalmological examination, her visual acuity was 20/400. Slit-lamp examination later confirmed diffuse cystine crystals throughout the corneal stroma. Other findings included astigmatism and bilateral hypopigmented retinopathy. The diagnosis of cystinosis was further confirmed by leukocyte cystine levels of 3.94 nmol half cystine/mg protein (normal, 0–0.3 nmol half cystine/mg protein).
Genetic testing was also performed. A positive homozygous exon 3 to 5 deletion in the cystinosin (CTNS) gene was found. This proved to be a novel mutation not previously described and was confirmed to be pathogenic for cystinosis.
The patient was treated for her coexisting urinary tract infection, started on electrolyte supplements, oral mercaptemine (cystagon) 90 mg four times daily and topical mercaptemine (cysteamine hydrochloride 0.55%) eye drops 6 times daily. Her systemic condition improved, and she was discharged after a month.
Figure 1
Use of two mobile phones to visualize corneal cystine crystals. A, Smartphone 1 (left) is used as the light source and smartphone 2 (right) captures a close-up image. B, Smartphone 1 is swiveled 30°-45° to obtain the specular reflection effect.
Figure 2
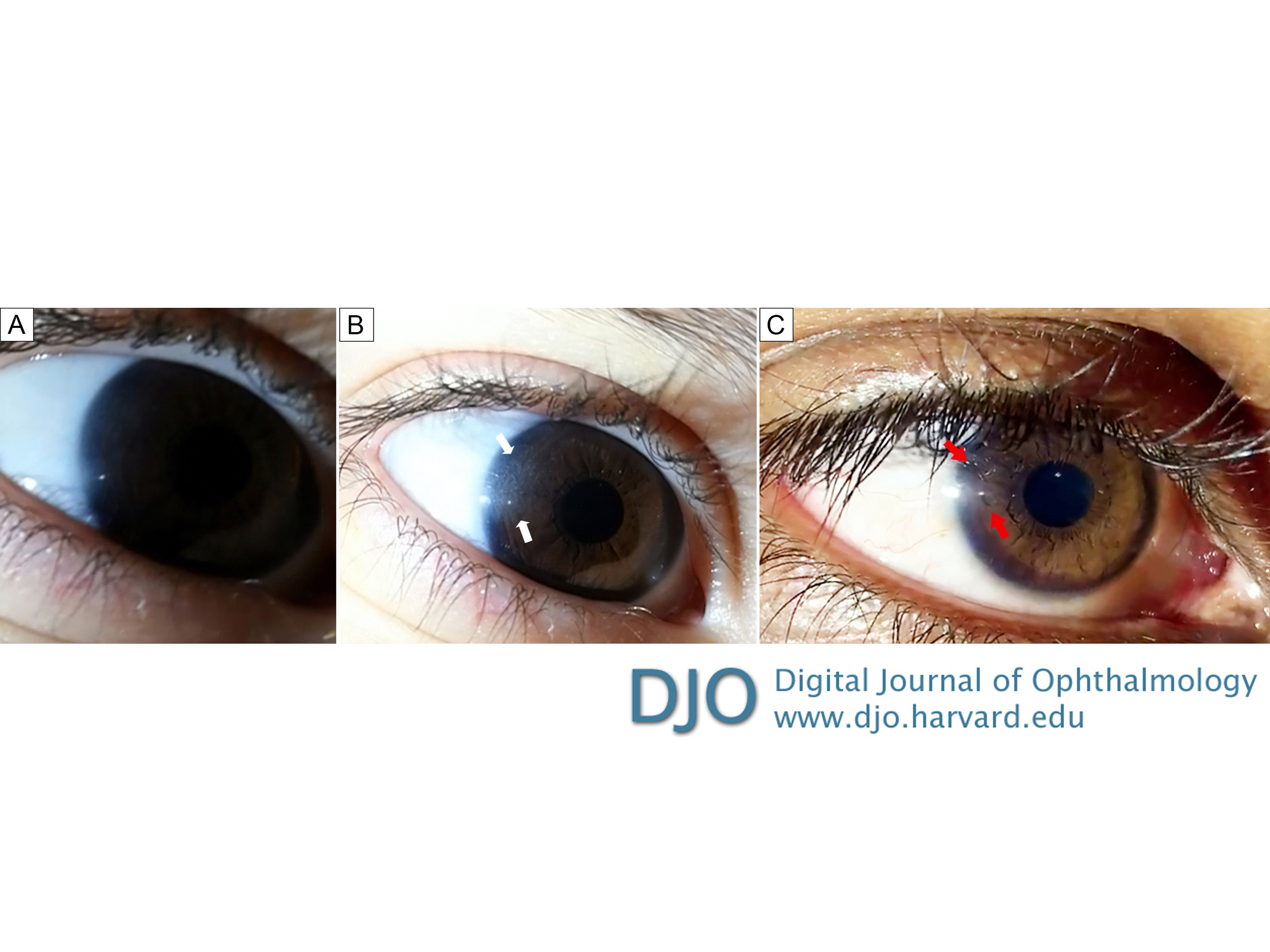
Video stills of the right corneal reflection during the maneuver. A, Normal-appearing cornea in the patient’s eye when using only a single smartphone to capture the image. B, With an additional smartphone used as a light source, the temporal area (white arrows) of the affected cornea shows the “ground-glass” haziness; this appearance can be visualized when the light source is “swiveled” in a position just temporal to the eye. C, Eye of an individual without cystinosis. Note the lack of peripheral “ground glass” haze adjacent to the corneal light reflex (red arrows).
Video 1
Video illustrating the technique using two smartphones to look for corneal cystine crystals.
Cystinosis is a rare metabolic condition affecting 1 in 180,000 individuals.(1) This autosomal recessive disease is a result of mutations in the 17p13.2 chromosome.(1,2) The changes in the CTNS gene cause a dysfunction or complete lack of production in the lysosomal membrane protein, cystinosin.(1,3) In the absence of a functional transporter, cystine cannot be recycled from within the lysosomes to the cytosol of the cell.(1,3) The low solubility of the molecule gives rise to crystal formation and accumulation within the lysosomes.(4) As a result, oxidative stress, cell injury, and apoptosis occur in cells throughout multiple organs.(4)
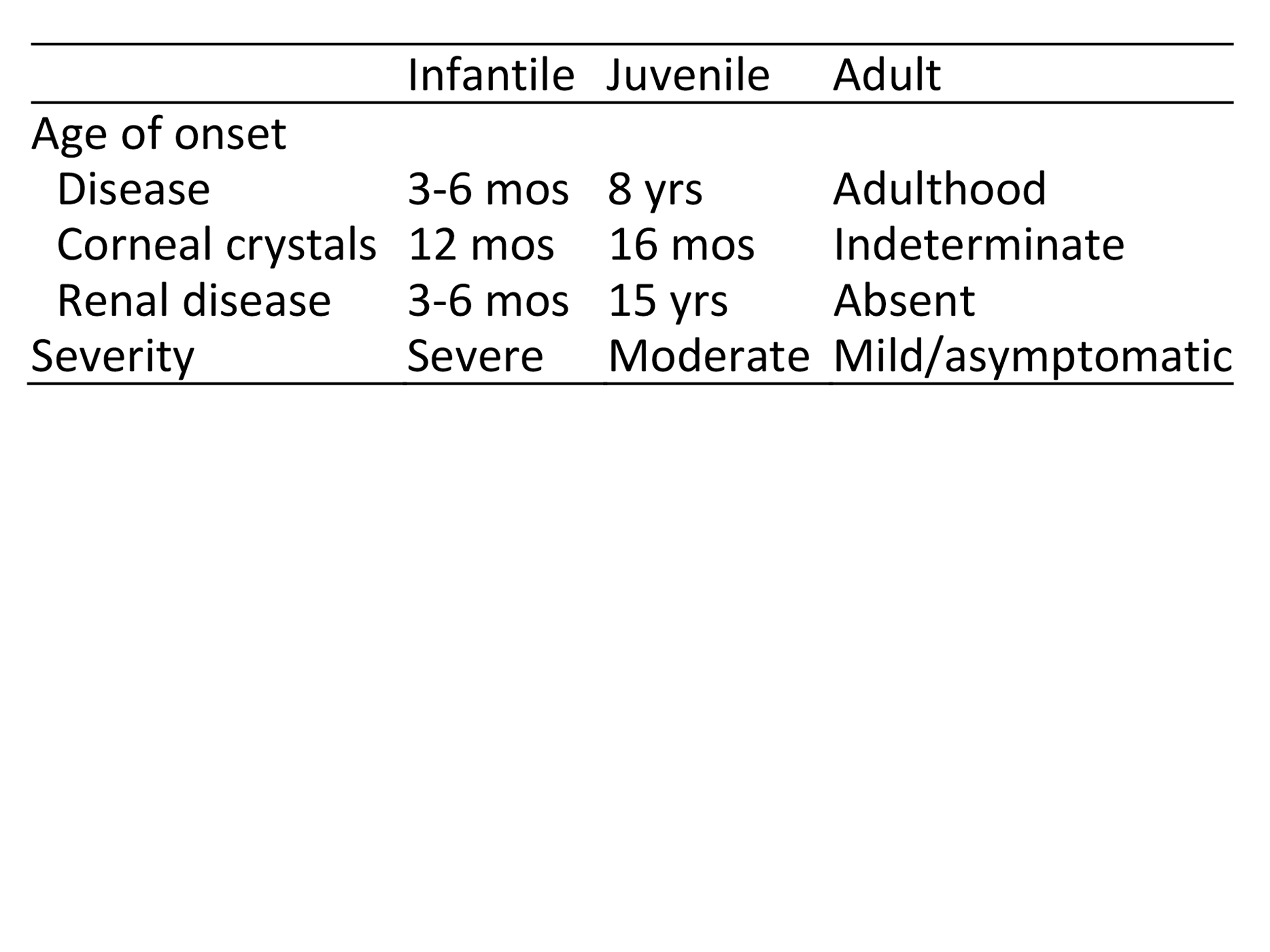
Based on the type and extent of the mutation, the condition can manifest in three forms relating to initial symptom onset—infantile (most common), juvenile, and adult (also known as ocular or non-nephropathic). See Table 1.
In infantile cystinosis, the first symptoms present as Fanconi syndrome, with hypokalemia, hyponatremia, metabolic acidosis, and hypophosphatemia.(1-3,5,6) In the eye, accumulation of cystine crystals occur in the corneal epithelium, stroma, and endothelium.(7) Deposits begin in the corneal peripheries and advance toward the center with age.(7) Over time, the iris, ciliary body, choroid, and optic nerve become involved.(7) Thus, ocular manifestations include photophobia, foreign body manifestation, blepharospasm, and pigmented retinopathy.(3,8)
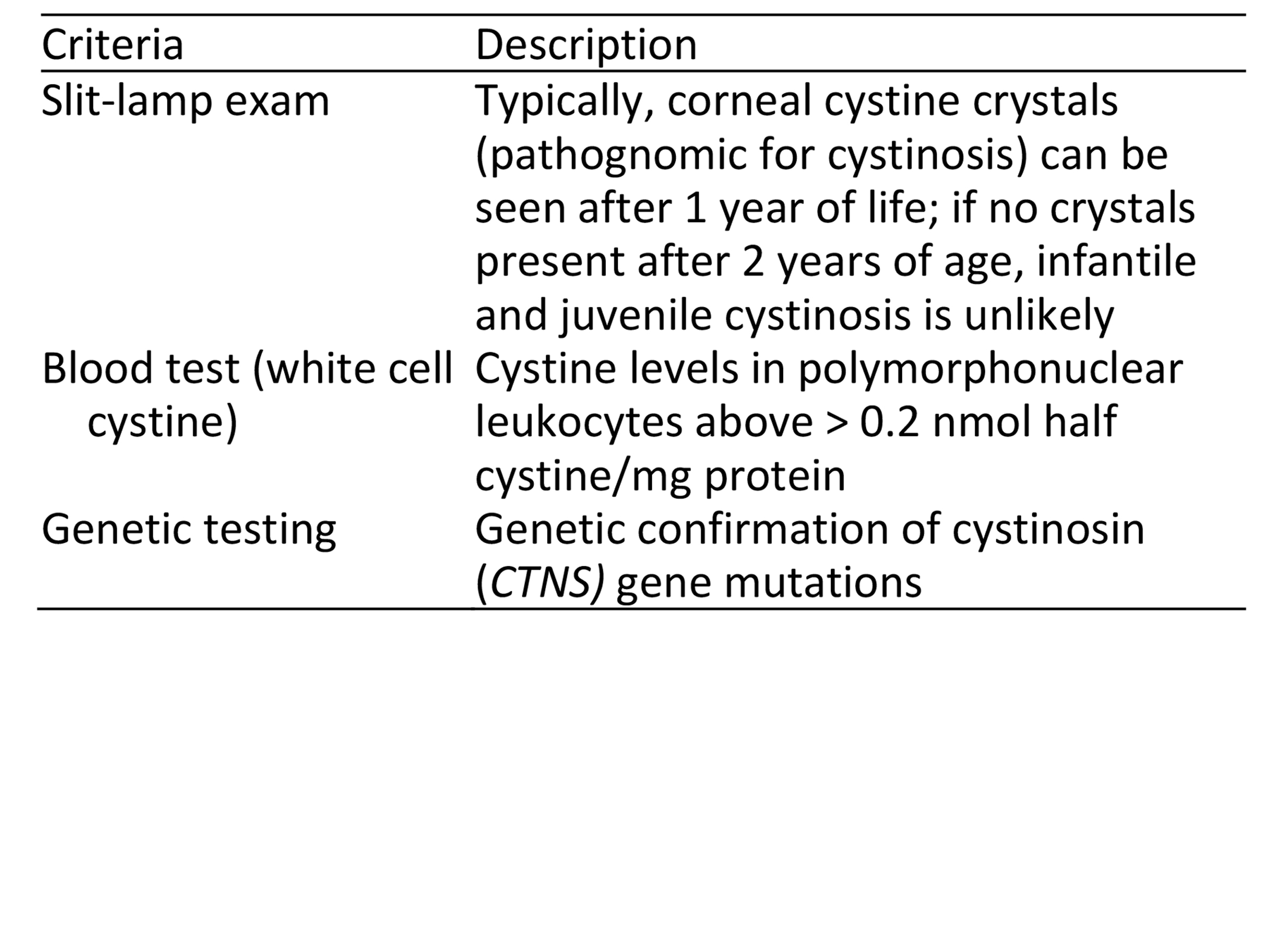
There are three ways to confirm cystinosis. This is based on the clinical detection of cystine crystal accumulation or confirming the underlying genetic cause. See Table 2.
Unaided, cystine crystals are not evident on examination and affected eyes are likely to appear normal. A slit-lamp examination looking for corneal deposits is a sensitive tool, but it is usually only available in ophthalmology clinics and used primarily by ophthalmic professionals. This report shows that it was possible to detect corneal findings using two smartphones—one for image capture, and the other as a light source capable of generating a specular effect. The bright white light of a smartphone appears to be optimum for this purpose; light produced from a pen flashlight or transilluminator is often not bright enough to generate the same effect. Also, a smartphone may be more readily available than a pen flashlight in any given setting, whether the clinician is in casualty, primary care, or the pediatric assessment unit. Furthermore, the high quality of modern smartphone cameras makes it possible to focus on the cornea and video record or capture an anterior segment photograph of adequate resolution with minimal practice. When the photograph is magnified, more corneal detail can be revealed.
Although slit-lamp examinations are the gold standard tool for monitoring corneal disease, smartphones can be successfully used as a screening tool and have clinical utility in increasing the pretest probability of cystinosis. Any healthcare professional who uses basic fundoscopy as a means of diagnosis, such as a general practitioner, pediatrician, or optometrist, will be able to appreciate the haziness and crystal accumulation. Furthermore, as ocular manifestations of infantile cystinosis are evident by 18 months of age,(9) screening beyond this age can greatly reduce the number of false negatives. However, false positive results are theoretically possible with other causes of crystalline keratopathies, including dystrophies (Schnyder corneal dystrophy, Bietti corneoretinal dystrophy), infections (Streptococcus viridans), and drug-related causes (ciprofloxacin and fluorometholone).(10)
Because smartphones are readily available and owned by virtually all clinicians, this technique could be useful for nonophthalmologists, especially those who work in remote and rural centers, without ready access to eye specialists. Through imaging evidence, a robust referral can be made to ophthalmologists instead of relying on only the clinical history of the disease. Moreover, there is value to this technique in countries that do not have molecular cystinosin studies or cystine level tests readily available, because they heavily depend on slit-lamp examinations for diagnosis.(11)
In conclusion, a “positive” finding will facilitate diagnostic testing, as demonstrated in this case. This helps to minimize further invasive and potentially stressful investigations in a child with nonspecific findings, especially when trying to pinpoint a metabolic process that is heterogeneous in etiology. A “negative” finding would theoretically reduce the likelihood of cystinosis. However, further studies are required to determine the sensitivity and specificity of this technique in detecting corneal crystals, with consideration of age and differing cystine crystal densities.
1. Elmonem MA, Veys KR, Soliman NA, Dyck Mv, Heuvel LPWJvd, Levtchenko E. Cystinosis: a review. Orphanet J Rare Dis 2016;11:47.
2. Turner NN, Lamiere N, Goldsmith DJ, et al. Oxford Textbook of Clinical Nephrology. 4th ed. New York: Oxford University Press; 2015.
3. Oshima RG, Willis RC, Furlong CE, Schneider JA. Binding assays for amino acids: The utilization of a cystine binding protein from Escherichia coli for the determination of acid soluble cystine in small physiological samples. J Biol Chem 1974;249:6033-9.
4. Gahl WA, Kuehl EM, Iwata F, Lindblad A, Kaiser-Kupfer MI. Corneal crystals in nephropathic cystinosis: natural history and treatment with cysteamine eyedrops. Mol Genet Metab 2000;71:100-120.
5. Fecarotta C, Huang W. Pediatric genetic disease of the cornea. J Pediatr Genet 2014;3:195-207.
6. Gahl WA, Balog JZ, Kleta R. Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med 2007;147:242-50.
7. Ecel M, Sari A, Delibaş A. Diagnosis of nephropathic cystinosis in a child during routine eye exam. Turk J Ophthalmol 2017;47:292-5.
8. Dufier JL, Dhermy P, Gubler MC, Gagnadoux MF, Broyer M. Ocular changes in long-term evolution of infantile cystinosis. Ophthalmic Paediatr Genet 1987;8:131-7.
9. Emma F, Nesterova G, Langman C, et al. Nephropathic cystinosis: an international consensus document. Nephrol Dial Transplant 2014;29:87-94.
10. Weiss JS, Khemichian AJ. Differential diagnosis of Schnyder corneal dystrophy. Dev Ophthalmol 2011;48:67-96.
11. Alcantara-Ortigoza MA, Martinez-Bernal AB, Belmont-Martinez L, et al. CTNS gene analysis emphasizes diagnostic value of eye examination in patients with cystinosis. J Pediatr Genet 2013;2:129-32.
 Welcome, please sign in
Welcome, please sign in