|
|
 |
|
|
|
|
|
Orbital desmoplastic small round cell tumor in an infant
Digital Journal of Ophthalmology
2018
Volume 24, Number 4
December 31, 2018
DOI: 10.5693/djo.02.2018.10.001
|
Printer Friendly
Download PDF |
|
|
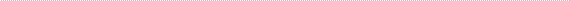 Amy Huang, BS
Amy Huang, BS | University of Central Florida College of Medicine, Orlando Nishita Patel, MD | Florida State University College of Medicine, Tallahassee, Florida
|
|
|
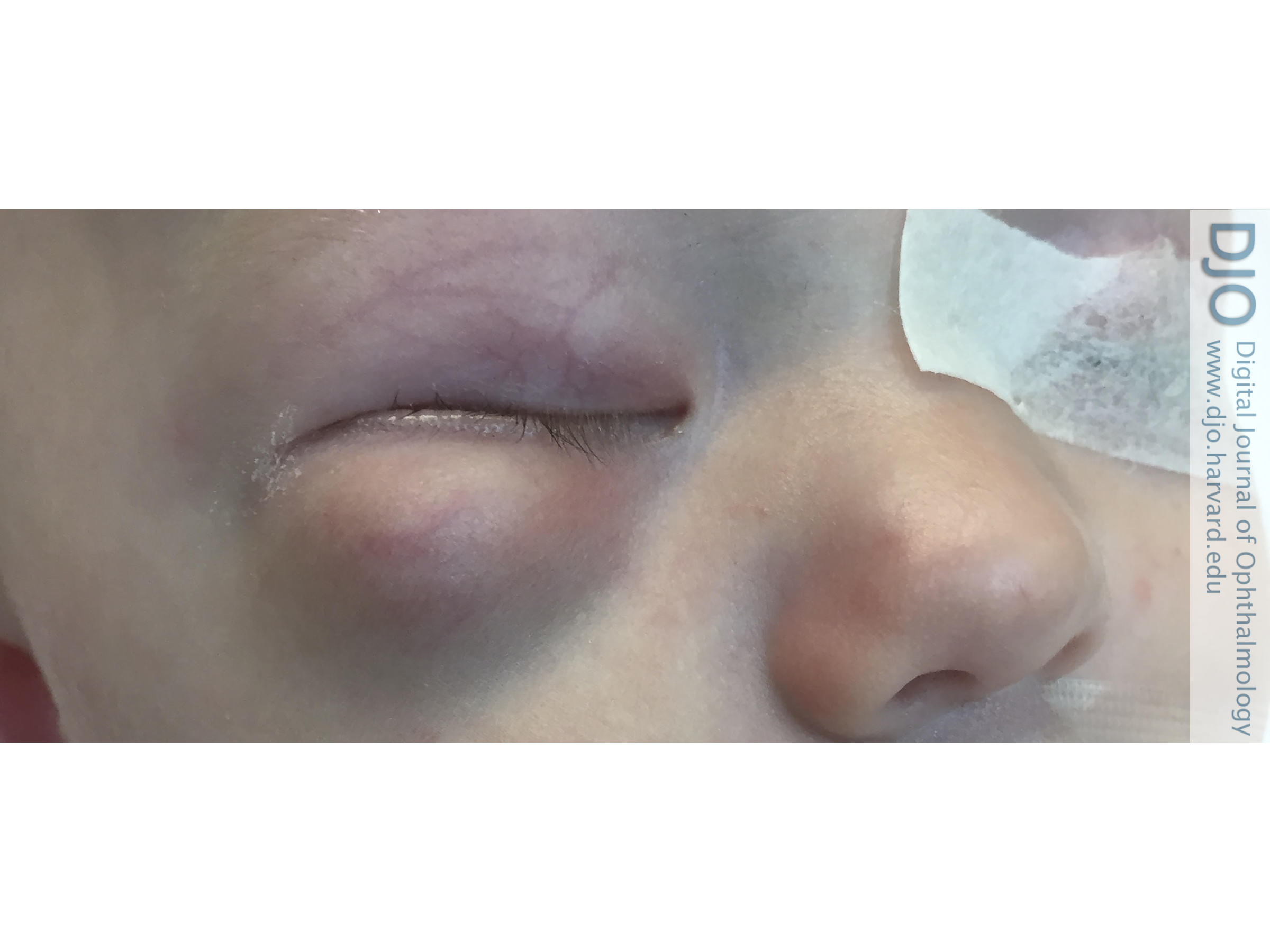
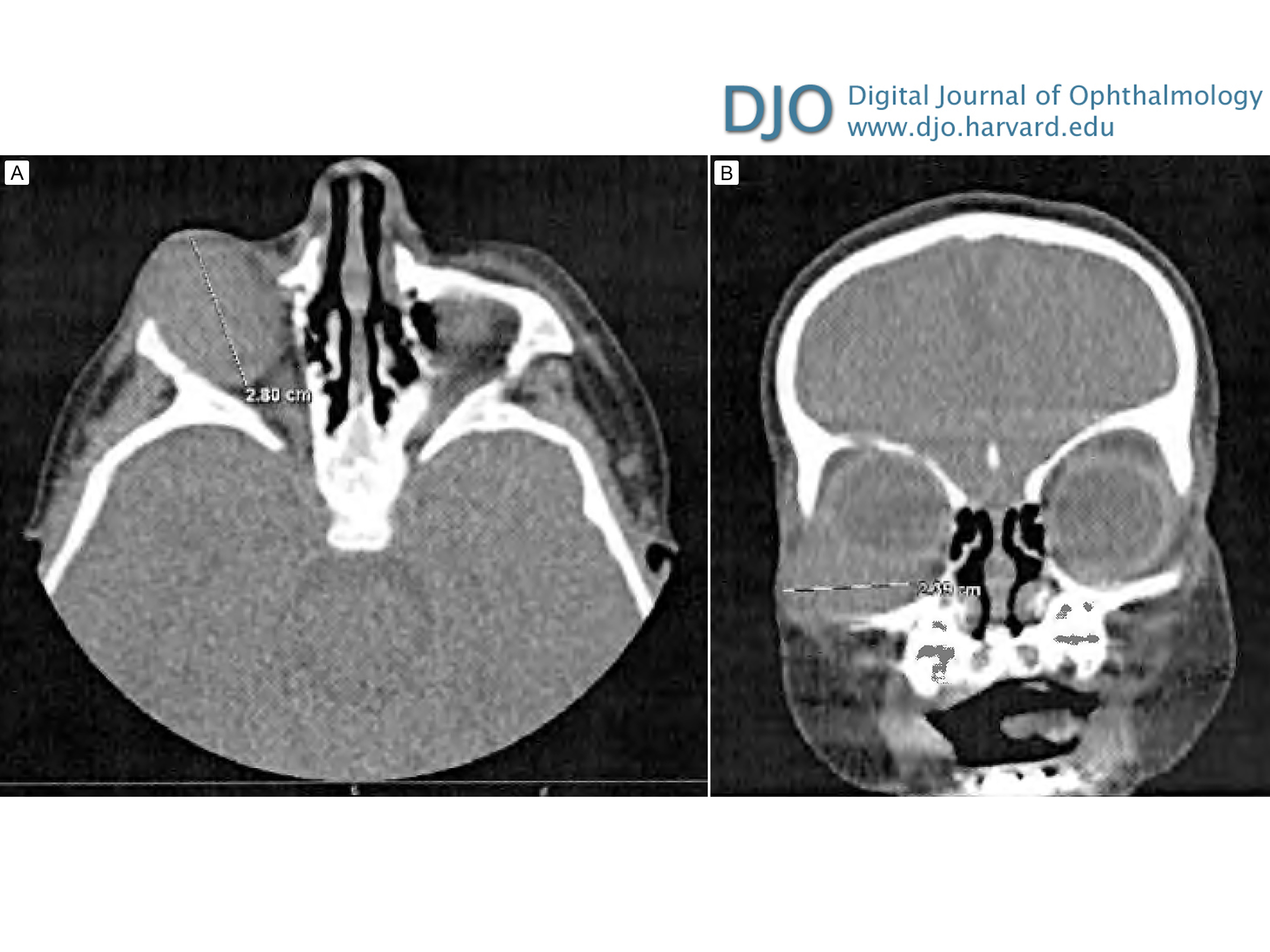
| Abstract | | Desmoplastic small round cell tumor (DSRCT) is a rare, aggressive malignancy that primarily involves the serosal surfaces of the abdomen and pelvis and has a poor prognosis. Orbital involvement is extremely rare. We report the case of a 2-month-old boy who presented with a right infraorbital mass consistent with a DSRCT and causing mass effect and superonasal globe displacement. To our knowledge, this is the first case of orbital DSRCT in an infant. | | | Introduction | | Desmoplastic small round cell tumor (DSRCT) is a rare, highly aggressive mesenchymal tumor that was first described by Gerald and Rosai in 1989.(1) Since its initial classification, only about 450 cases have been reported.(2) DSRCT most commonly affects adolescent or young adult males, with the median age of diagnosis ranging between 14 and 26 years.(3-6) These tumors tend to involve the serosal surfaces of the abdomen and pelvis, and patients typically present with widespread peritoneal involvement.(5,7) Extra-abdominal DSRCT, especially in the head and neck, is rare. The diagnosis of DSRCT is based on both immunohistologic and cytogenic findings. Generally tumors show small, round, or spindled cells lying in a desmoplastic stroma.(5) The hallmark of this tumor is a t (11;22) (p13;q12) translocation.(7) Although this translocation is unique to DSRCT, these two chromosomal regions are also associated with other malignant developmental tumors: 22q12 is the site of the Ewing sarcoma gene,(8) EWS, and 11p13 is the site of the Wilms tumor gene, WT1.(9) DSRCT has an extremely aggressive clinical course and a very poor prognosis. Even with aggressive multimodal treatment, median survival ranges from 17 to 25 months, with fewer than 20% of patients surviving beyond 5 years.(10) | | | Case Report | A previously healthy 2-month-old boy presented at Halifax Health, Daytona Beach, Florida, with a 3-day history of swelling inferior to his right orbit (Figure 1). His parents reported that his overhead mobile fell and struck him on the face while sleeping. Computed tomography (CT) of the facial bones without contrast showed a large 2.8 × 2.4 cm “hematoma” in the right orbit, inferior to the globe, causing mass effect and superior medial displacement of the globe, without any evidence of fracture (Figure 2). Ophthalmology was consulted, and physical examination showed a large, firm protrusion inferior to the right eye, and a normal pupil, intraocular pressure, and optic nerve. Because the patient failed to improve, he underwent drainage of the right orbital lesion. During the procedure, firm tissue was seen instead of blood, and biopsies of the tumor were submitted for histopathological evaluation.
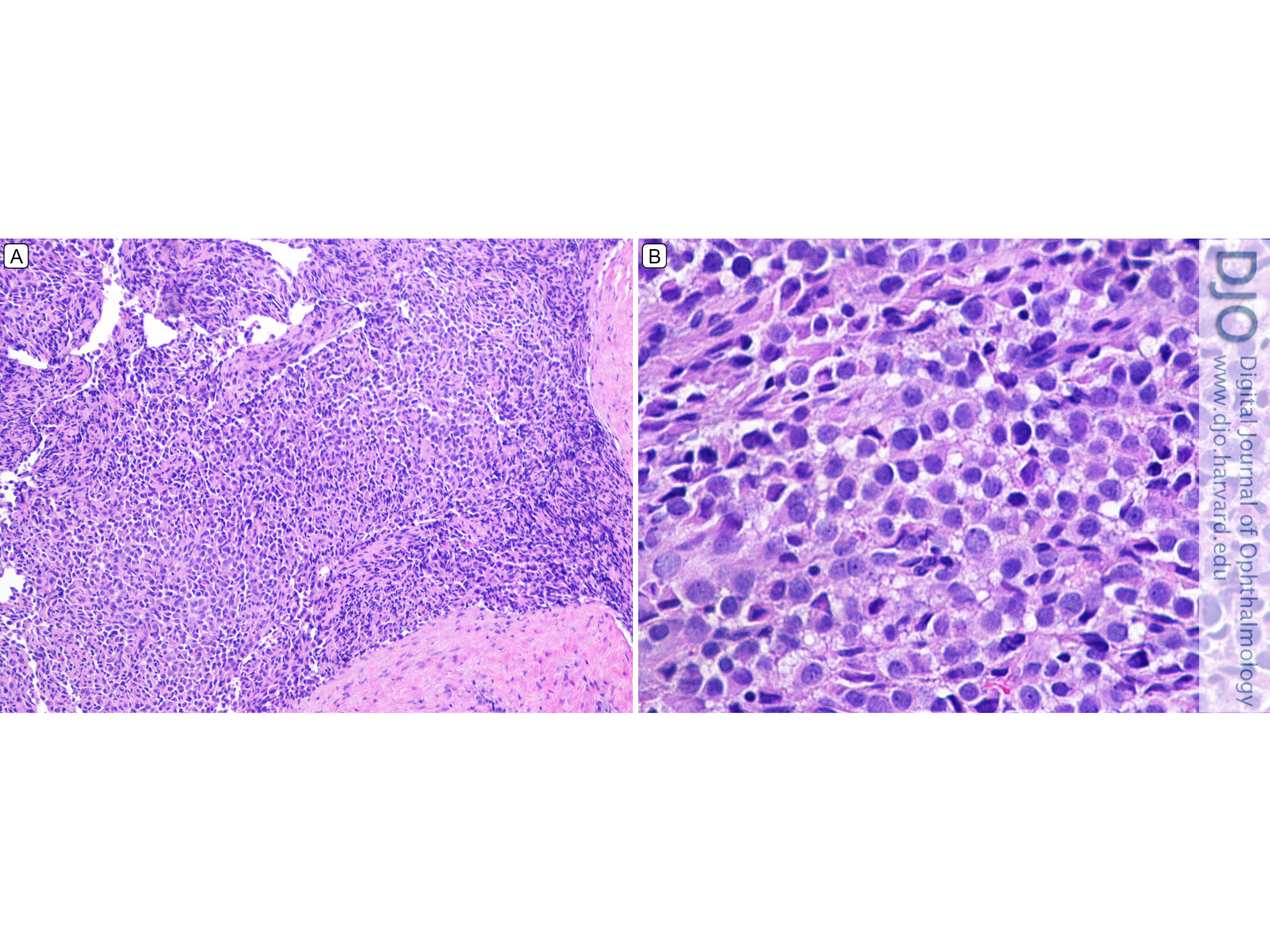
Hematoxylin-eosin sections of tumor tissue revealed a tumor composed of small round blue cells infiltrating the surrounding desmoplastic stroma (Figure 3). Immunohistochemical staining revealed tumor cells that stained positive for CD99 and scattered cells that stained positive for desmin, MyoD1, and WT1. Staining was negative for synaptophysin, AE1/3, CK OSCAR, CD45, S100, HMB-45, Melan A, Myf-4, and Tdt. Because this staining pattern did not point to a definitive diagnosis, specimens were sent for additional cytogenetic testing. Comprehensive genomic profiling demonstrated that the tumor was most consistent with DSRCT.
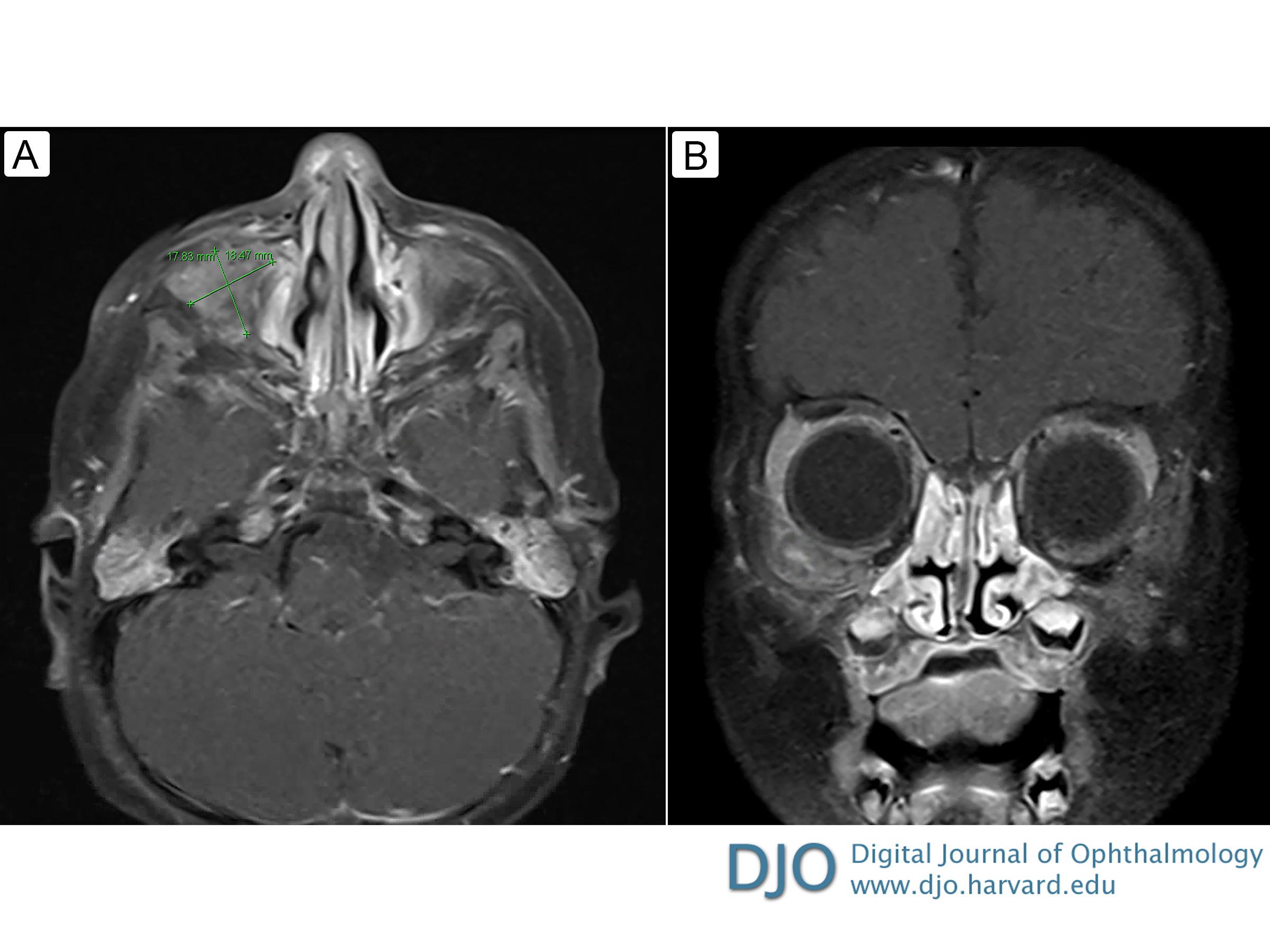
The patient also began to have increasing medial displacement of the right globe. Forced ductions of the right eye were normal, but extraocular motility was limited inferiorly and laterally due to mass effect of the tumor. Magnetic resonance imaging (MRI) of the head revealed a 3.3 × 2.2 × 2.1 cm right orbital extraconal and intraconal mass encasing and compressing the lateral rectus muscle, with mass effect on the right globe, inferior rectus muscle, and intra-orbital optic nerve segment (Figure 4). No intracranial metastases were noted.
CT of the abdomen and pelvis demonstrated metastatic disease as evidenced by lymphadenopathy, 3 mm right middle lung and lingular nodules, and a 2.0 × 1.2 cm hypoenhancing splenic mass. Bilateral bone marrow biopsies and a bone scintigraphy were negative. The patient was started on the Ewing sarcoma chemotherapy protocol AEWS0031, regimen B (dose-compressed arm with alternating vincristine-doxorubicin-cyclophosphamide and ifosfamide-etoposide cycles).(11) To avoid excessive radiation and orbital exenteration, a multidisciplinary tumor board agreed to initially proceed with chemotherapy without radiation and evaluate for localized surgery after at least 12 weeks of treatment. Repeat MRI 6 months later demonstrated a smaller 1.7 × 1.8 × 0.8 cm (previously 3.3 × 2.2 × 2.1 cm) mass with minimal mass effect on the right globe and inferior rectus muscle (Figure 5). Both right and left globes and optic nerves appeared normal. Repeat CT of the abdomen and pelvis demonstrated no appreciable lymphadenopathy, resolution of the splenic lesion, and no evidence of metastatic disease. Since initial diagnosis, the patient has completed 14 cycles of chemotherapy. Repeat MRI at 1-year follow-up demonstrated a stable orbital mass with normal appearance of right and left globes and optic nerves. | |
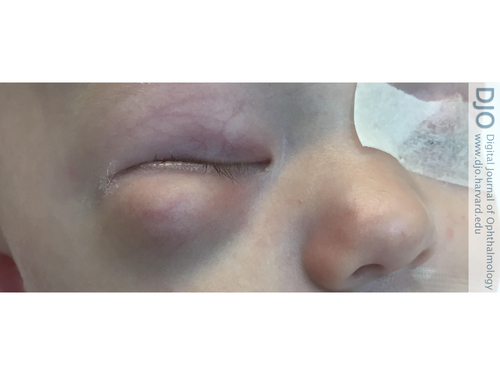
Figure 1
Right inferior orbital swelling in a 2-month-old boy on presentation.
|
|
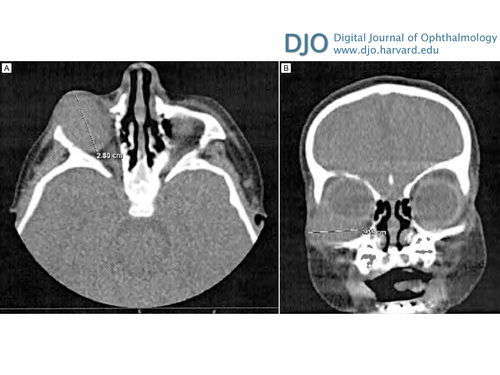
Figure 2
An inferolateral right orbital mass causing mass effect and superior displacement of the globe seen on axial (A) and coronal (B) contrast-enhanced computed tomographic images.
|
|
Figure 3
Histopathologic section of surgical specimen (hematoxylin-eosin). A, Low-power photomicrograph (original magnification ×40). B, High-power photomicrograph (original magnification ×100).
|
|
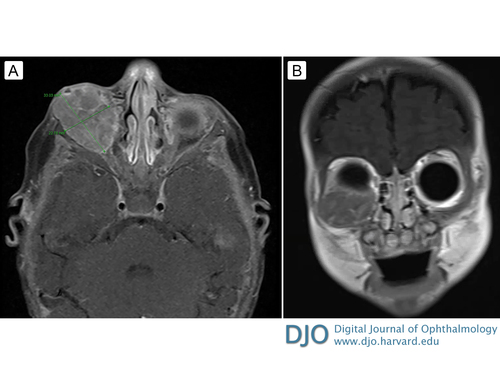
Figure 4
A, Axial T1-weighted magnetic resonance image (MRI) with contrast before chemotherapy. B, Coronal T1-weighted MRI with contrast before chemotherapy.
|
|

Figure 5
A, Axial T1-weighted MRI with contrast after chemotherapy. B, Coronal fat-suppressed T1-weighted MRI with contrast after chemotherapy.
|
|
| Discussion | DSRCT is a rare, aggressive malignancy that usually involves the serosal surfaces of the abdomen and pelvis in adolescent and young adult males. DSCRT in the head and neck is extremely uncommon. To date, there have been 13 documented cases of DSRCT involving the head and neck and only 3 documented cases involving the orbit.(12,13) Yoon et al described a left orbital mass in a 32-year-old man with visual disturbances, and eventual proptosis and vision loss.(14) Cobanoglu et al described a right orbital mass in a 4-year-old boy with ptosis, mild proptosis, and mass effect on the lateral rectus, inferior rectus, and optic nerve.(12) He et al described a left orbital mass in a 16-year-old boy with painful swelling and a palpable infraorbital mass.(13) To our knowledge, this is the fourth case of orbital DSRCT described in literature and the first case in an infant.
In the present case, several features are consistent with DSRCT. Immunohistologic revealed irregular sheets of small, round, blue cells infiltrating demosplastic stroma and cells positive for CD99, desmin, MyoD1, and WT1. Additionally, the comprehensive genomic testing revealed that the tumor was consistent with DSRCT. However, the manifestation of DSRCT in this patient is unusual because of both orbital involvement and the very young age of the patient.
Regardless of tumor location, there is no standard approach to treatment of DSRCT. Due to the rarity of this tumor, studies on various treatment modalities and their effect on survival are limited. Treatment options include surgery, chemotherapy, and radiation, but there is no standard approach to treatment.(15) Despite the use of aggressive multimodal treatment modalities, such as the combination of chemotherapy, radiation therapy, and surgery, the overall 5-year survival remains less than 20%. Because of our patient’s age and the location of the tumor, radiation therapy was not used, and the option of localized surgery was postponed for later evaluation.
DSRCT is a rare, aggressive malignancy. The diagnosis is based on clinical presentation and unique immunohistologic and genetic characteristics. Involvement of the orbit is extremely rare, but DSRCT should be included in a differential diagnosis for an orbital mass. Chemotherapy was an effective treatment in the present case.
Literature Search
PubMed was searched on April 1, 2018, without date restriction, for English-language articles, using the following terms: orbital desmoplastic small round cell tumor, desmoplastic small round cell tumor, and infant.
| | | References | 1. Gerald WL, Rosai J. Case 2. Desmoplastic small cell tumor with divergent differentiation. Pediatr Pathol 1989;9:177-83.
2. Honore C, Amroun K, Vilcot L, et al. Abdominal desmoplastic small round cell tumor: multimodal treatment combining chemotherapy, surgery, and radiotherapy is the best option. Ann Surg Oncol 2015;22:1073-79.
3. Bisogno G, Roganovich J, Sotti G, et al. Desmoplastic small round cell tumour in children and adolescents. Med Pediatr Oncol 2000;34:338-42.
4. Schwarz RE, Gerald WL, Kushner BH, Coit DG, Brennan MF, La Quaglia MP. Desmoplastic small round cell tumors: prognostic indicators and results of surgical management. Ann Surg Oncol 1998;5:416-22.
5. Lae ME, Roche PC, Jin L, Lloyd RV, Nascimento AG. Desmoplastic small round cell tumor: a clinicopathologic, immunohistochemical, and molecular study of 32 tumors. Am J Surg Pathol 2002;26:823-35.
6. Hassan I, Shyyan R, Donohue JH, et al. Intraabdominal desmoplastic small round cell tumors: a diagnostic and therapeutic challenge. Cancer 2005;104:1264-70.
7. Gerald WL, Ladanyi M, de Alava E, et al. Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round-cell tumor and its variants. J Clin Oncol 1998;16:3028-36.
8. Zucman J, Delattre O, Desmaze C, et al. EWS and ATF-1 gene fusion induced by t(12;22) translocation in malignant melanoma of soft parts. Nat Genet 1993;4:341-5.
9. Bonetta L, Kuehn SE, Huang A, et al. Wilms tumor locus on 11p13 defined by multiple CpG island-associated transcripts. Science 1990;250:994-7.
10. Dufresne A, Cassier P, Couraud L, et al. Desmoplastic small round cell tumor: current management and recent findings. Sarcoma 2012;2012:714986.
11. Womer RB, West DC, Krailo MD, et al. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 2012;30:4148-54.
12. Bengu Cobanoglu H, Hanna EY, Bell D, Esmaeli B. Desmoplastic small round cell tumor presenting as an ocular mass: unusual localization and remarkable surgical approach. Curr Oncol Rep 2017;19:80.
13. He XR, Liu Z, Wei J, Li WJ, Liu T. Primary desmoplastic small round cell tumor in the left orbit: a case report and literature review. Int Ophthalmol 2018.
14. Yoon M, Desai K, Fulton R, et al. Desmoplastic small round cell tumor: a potentially lethal neoplasm manifesting in the orbit with associated visual symptoms. Arch Ophthalmol 2005;123:565-7.
15. Kallianpur AA, Shukla NK, Deo SV, et al. Updates on the multimodality management of desmoplastic small round cell tumor. J Surg Oncol 2012;105:617-21.
| |
|
|
|
|
|
|
 Welcome, please sign in
Welcome, please sign in