|
|
 |
|
|
|
|
|
Optical coherence tomography findings and successful repair of retina detachment in Knobloch syndrome
Digital Journal of Ophthalmology
2017
Volume 23, Number 1
March 12, 2017
DOI: 10.5693/djo.02.2017.01.002
|
Printer Friendly
Download PDF |
|
|
Nazanin Ebrahimiadib, MD | Retina Service, Department of Ophthalmology, Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, Massachusetts Bobeck S. Modjtahedi, MD | Retina Service, Department of Ophthalmology, Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, Massachusetts Kevin Ferenchak, MD | Retina Service, Department of Ophthalmology, Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, Massachusetts Thanos Papakostas, MD | Retina Service, Department of Ophthalmology, Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, Massachusetts Jason S. Mantagos, MD | Department of Ophthalmology, Boston Children’s Hospital, Harvard Medical School, Boston, Massachusetts Demetrios G. Vavvas, MD, PhD | Retina Service, Department of Ophthalmology, Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, Massachusetts
|
|
|
| Abstract | | A 7-year-old Afghani girl was referred to the retina clinic of Massachusetts Eye and Ear for a chronic-appearing, macula-off retinal detachment in the left eye. On examination, best-corrected visual acuity was 20/400 in the right eye and 20/800 in the left eye. She had bilateral horizontal nystagmus. Ophthalmoscopy revealed prominent choroidal vessels, chorioretinal atrophy in the macular area, attenuated retinal vasculature, and pale optic discs bilaterally. Spectral domain optical coherence tomography demonstrated atrophy of the choriocapillaris and the retinal pigment epithelium, retinal thinning, and abnormal foveal contour. In the right eye, findings were reminiscent of dome shape maculopathy with an adjacent lesion suspicious for inactive choroidal neovascularization. A suspected diagnosis of Knobloch syndrome was confirmed by genetic testing, which showed a homozygous variant in exon 33 of the COL18A1 gene defined as c.3213dupC. She underwent cryotherapy and scleral buckling surgery in the left eye and remained attached bilaterally at 3 years’ follow-up, with progressive myopia and best-corrected visual acuity of 20/100 in the right eye and 20/125 in the left eye. | | | Introduction | Knobloch syndrome (KNO) is a rare autosomal recessive disorder, first described in 1971 in 5 siblings, characterized by high myopia (−10 to −20 D), vitreoretinal degeneration with retinal detachment, and occipital encephalocele.(1) It is caused by homozygous or compound heterozygous mutation in the COL18A1 gene leading to deficiency in collagen XVIII. Additional ocular manifestations include white fibrillar vitreous condensation, diffuse severe vitreoretinal atrophy, persistent fetal vasculature, distinct macular atrophy, cone–rod dysfunction, electroretinogram diminution, retinal pigment epithelial atrophy, prominent choroidal vessels, smooth (cryptless) irides, zonular weakness with temporal lens subluxation, pigment dispersion syndrome and associated glaucoma, and some degree of cataract.(2) Neurologic deficits include epilepsy, developmental delay, and midline cranial defects. Occipital defects may range from bony defects to an encephalocele.(3) We report spectral domain optical coherence tomography (SD-OCT) findings in a patient with KNO syndrome and successful retinal detachment repair.
| | | Case Report | A 7-year-old Afghan girl with high myopia (−10.00 D), seizures, micropolygyria, and retinal degeneration was referred to the Retina Clinic of Massachusetts Eye and Ear for a chronic retinal detachment in her left eye detected on routine eye examination. She had a full full-field electroretinogram at 2 months old of age, with readily detectable responses in both scotopic and photopic conditions. Given her clinical presentation, the diagnosis of KNO syndrome was suspected, and the patient was referred for genetic testing. She had no gross defect in the pattern of hair growth on her head. Genetic testing revealed that the patient was homozygous in exon 33 of the COL18A1 gene for a sequence variant defined as c.3213dupC, which is consistent with the diagnosis of KNO syndrome.
Her visual acuity was 20/400 in the right eye and 20/800 in the left eye. She exhibited horizontal conjugate nystagmus. Tensions were normal in each eye. Anterior segment structures in the right eye were unremarkable on slit-lamp examination. There were posterior synechiae present in the left eye. Irides were normal. Dilated fundus examination revealed areas of optically empty vitreous, slightly tilted optic discs, focal patches of chorioretinal atrophy in macular area, posterior staphylomas, attenuated arteries, and widespread pigmentary changes bilaterally. The left eye had a temporal circular patch of depigmentation and an inferior retinal detachment with a small tear inferotemporally (Figure 1A,B). B-scan ultrasonography confirmed the retinal detachment (Figure 1C). Axial lengths measured as 26.8 mm in the right eye and 24.3 mm in the left eye. SD-OCT confirmed the presence of a macula-off retinal detachment (Figure 1D). The patient underwent scleral buckle surgery with placement of a 275 encircling band, cryotherapy, and anterior chamber paracentesis with synechiolysis in the left eye. The patient had an uneventful postoperative course. At 3 years’ follow-up, her retina remained attached, and best-corrected visual acuity was 20/100 in the right eye and 20/125 in the left eye.
Follow-up SD-OCT examinations revealed thin retinae and lack of normal foveal contour bilaterally. The choroid was not overtly thin and there was a striking appearance of large choroidal vessels. There was a mound of elevated tissue and a subretinal lesion with adjacent retinal lucencies within the macular staphyloma in the right eye (Figure 2). | |
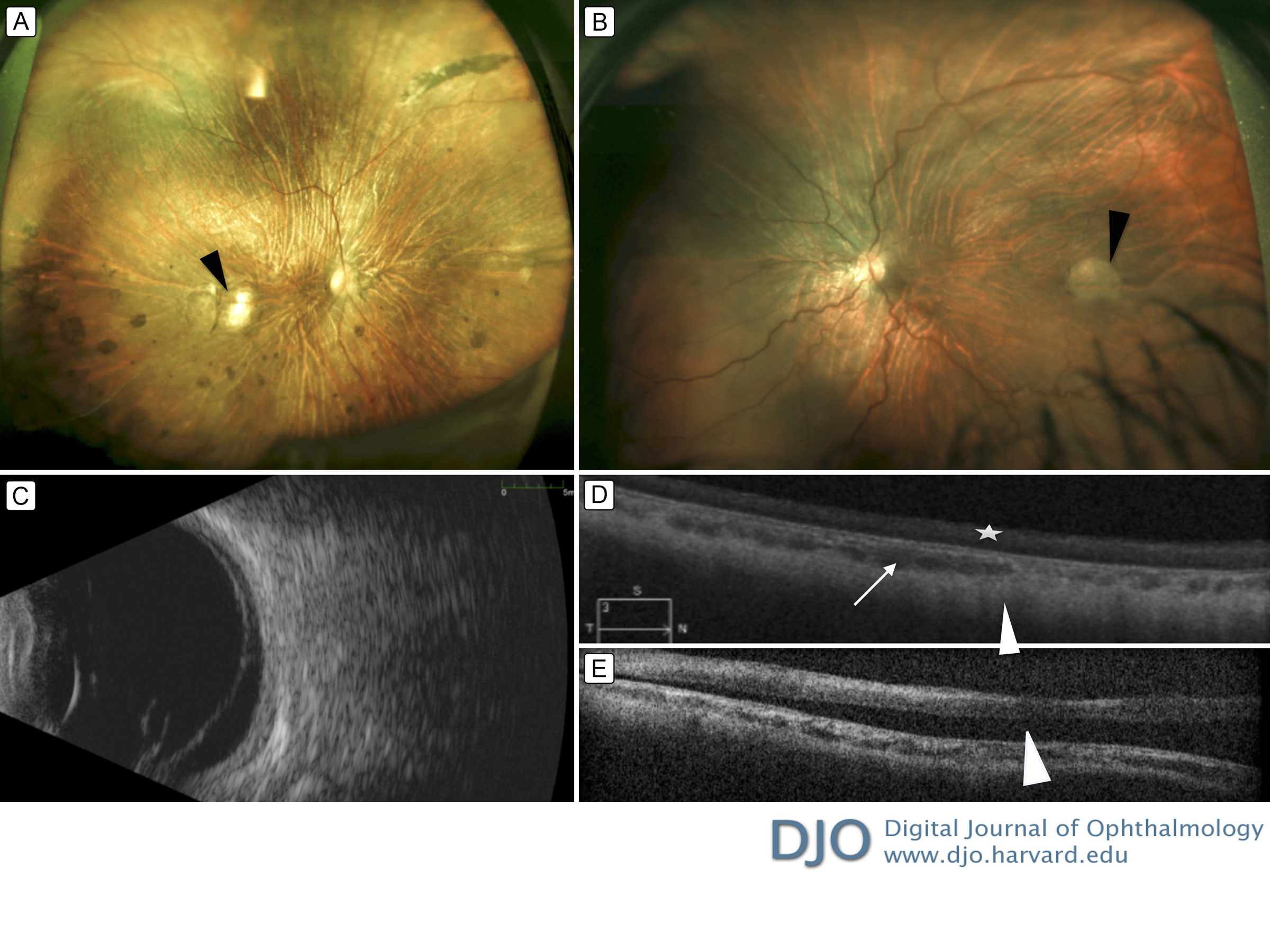
Figure 1
Widefield fundus photograph, B scan, and spectral domain optical coherence tomography (SD-OCT) at initial presentation. A, Fundus photograph of the right eye showing areas of optically empty vitreous, slightly tilted pale optic disc, posterior staphyloma, attenuated arteries, widespread pigmentary changes, and a focal choroioretinal atrophy (black arrowhead) at the inferotemporal part of the macula. B, Fundus photograph of the left eye showing areas of optically empty vitreous, slightly pale optic disc, a patch of chorioretinal atrophy at the temporal macula (black arrowhead), posterior staphyloma, attenuated arteries, and an inferior retinal detachment, with no a tear inferotemporally. C, B-scan ultrasonography of the left eye demonstrating a membrane consistent with retinal detachment. D, SD-OCT scans through the fovea of the right eye revealing prominent choroidal vessels (white arrow) and atrophic retina, retinal pigment epithelium, and choriocapillaris. There is a homogenous hyper reflectivity in the internal retinal layers (white star) and a lack of the normal foveal contour. The outline of the sclera is partially visible (white arrowhead). E, SD-OCT through the fovea of the left eye shows subretinal fluid beneath the fovea (white arrowhead).
|
|
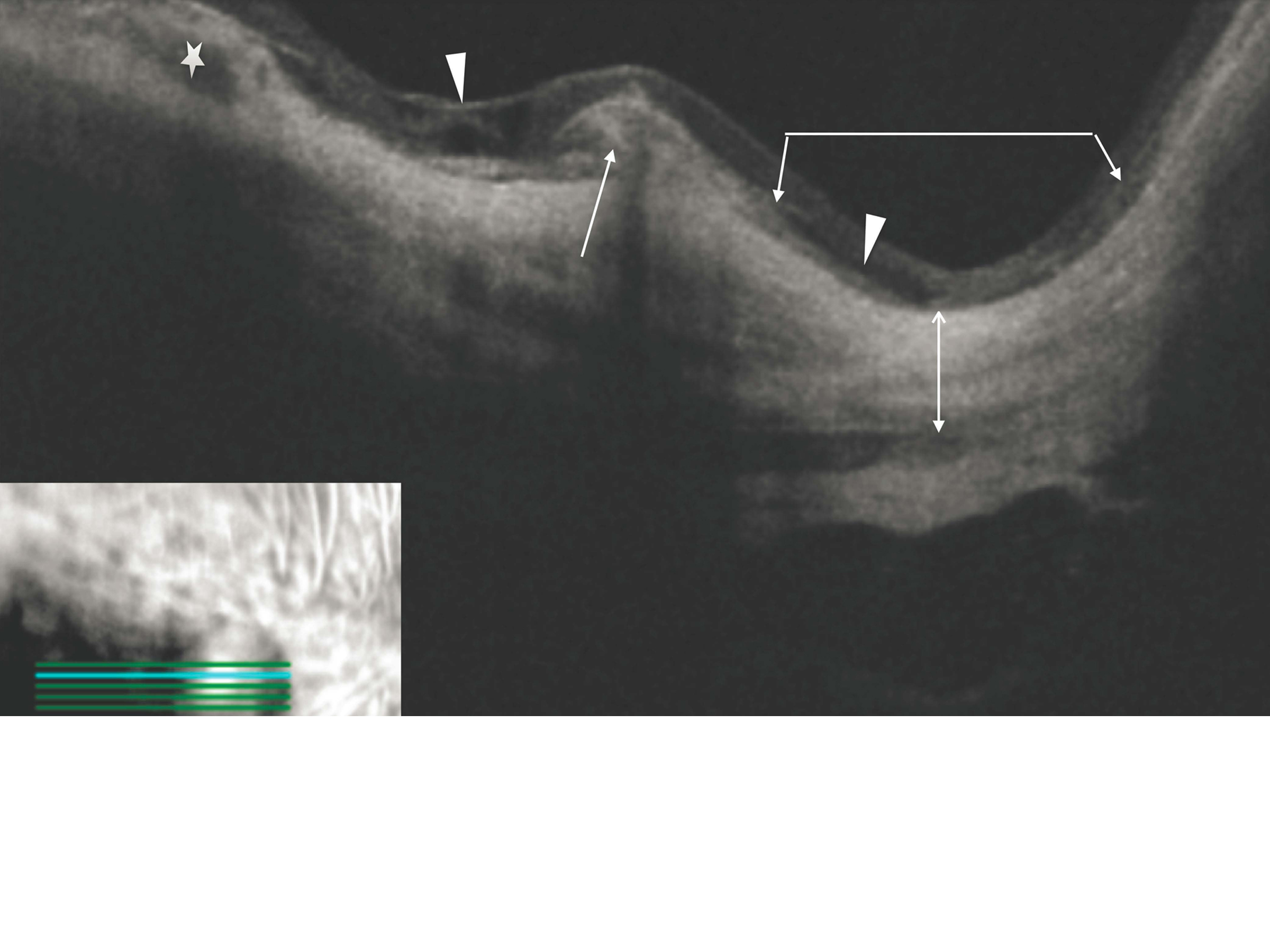
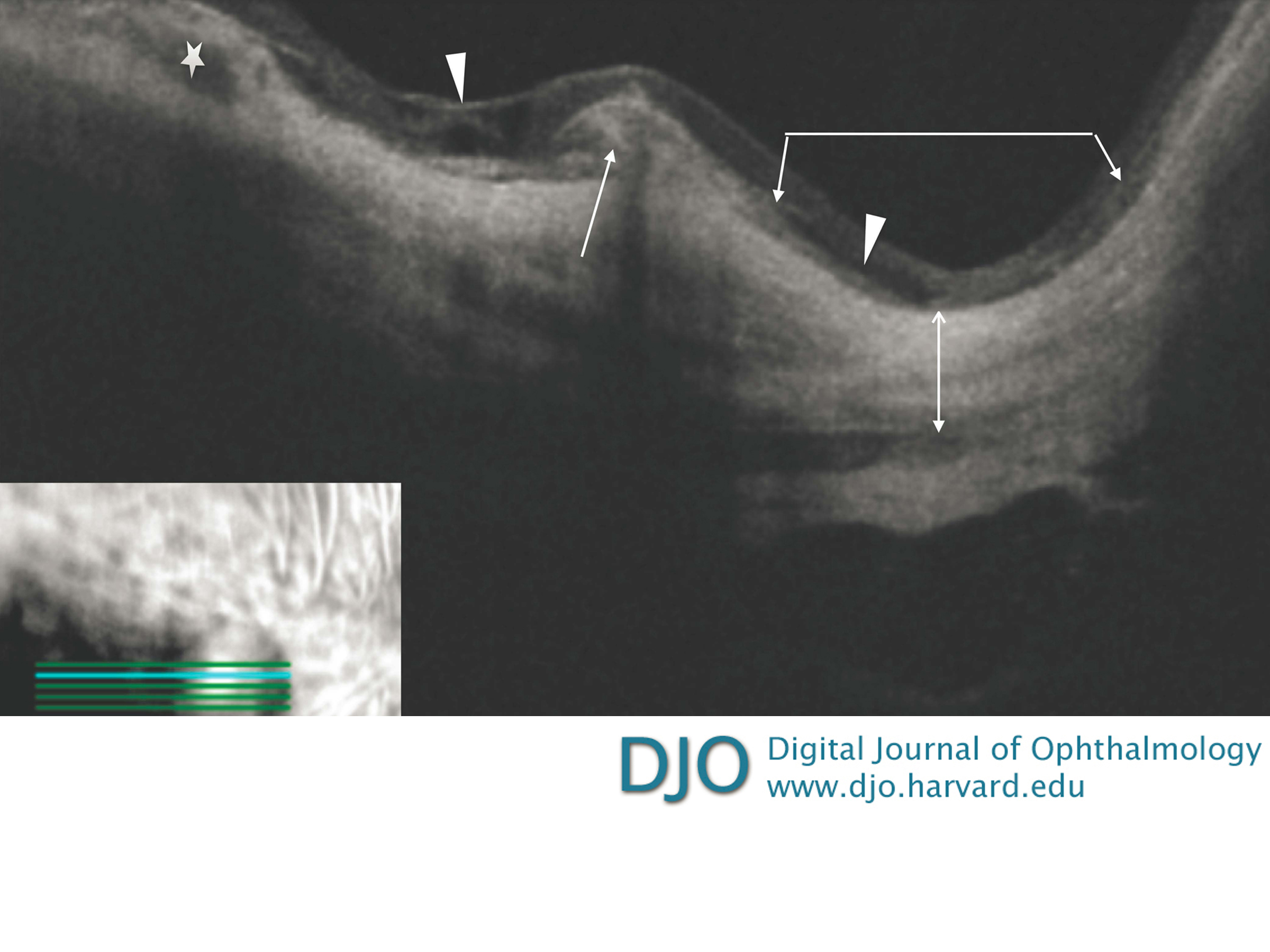
Figure 2
Spectral domain optical coherence tomography image of the right macula next to depigmented chorioretinal and retinal pigment epithelial atrophy. There is an elevated subretinal lesion without intra- or subretinal fluid, thus suspicious for inactive choroidal neovascularization (white arrow). The adjacent retina shows schisis (white arrowheads). The right side of the image shows a staphyloma (the area under the angled line with two arrowheads). The choroid is atrophic in this section and is only visible in the left part of the image (white star). The scleral outline (line with two arrowheads) can be seen very clearly, and there is an area reminiscent of dome-shaped maculopathy.
|
|
| Discussion | Mutations in the COL18A1 gene, which codes for three isoforms of collagen XVIII in the retina, are causative for Knobloch syndrome. Collagen XVIII is thought to affect the function of ocular basement membranes and participates in vitreous attachment to the inner limiting membrane. It is also expressed in the retinal pigment epithelium (RPE), Bruch’s membrane, and retinal and choroidal vasculature.(4)
OCT analysis of the fundus of our patient revealed a stable appearance over 3 years; prominent choroidal vessels and relative attenuation of the RPE and choriocapillaris signal.
The neurosensory retina was thin with altered reflectivity of the layers, which were difficult to discern. The right sclera showed changes that resembled dome-shaped maculopathy, seen in myopic staphylomas, despite the lack of a very elongated globe. In addition, there was an elevated subretinal lesion with adjacent retinal lucencies that could potentially represent a chorioretinal scar from old choroidal neovascularization. Adjacent to this part, the retina showed schisis changes that appeared as lucent or hyporeflective cavities. Most strikingly, multiple scans failed to demonstrate the presence of a foveal pit or normal foveal contour, consistent with Knobloch’s original description of foveal hypoplasia.(1)
Axial length cannot completely explain the degree of myopia in our patient, especially in the left eye. It is likely that keratometric or crystalline changes contributed to the degree of myopia in our case. However, AlBakri et al recently reported 7 patients with Knobloch syndrome whose axial lengths were consistent with axial myopia.(5) Thus, the etiology of the myopia in each case may vary.
Although vitreoretinal degeneration is usually the prominent feature in many patients with KNO, our Afghani patient had prominent chorioretinal degeneration, similar to a report in an Iranian family with KNO.(6)
Older patients with KNO syndrome have been reported to show a high incidence of phthisis bulbi as a result of failed repair of retinal detachment, diagnosis and repair of which in patients with KNO can be made difficult by miotic pupils and ectopic lenses. Visual prognosis, even after surgery, is poor, with at least one eye often progressing to phthisis bulbi or blindness.(7) Most reported cases require more than a single surgery. Moysidis et al reported that 11 of 12 patients (92%) with KNO and retinal detachment who underwent repair with vitrectomy or scleral buckle had a final visual acuity of 20/200 or worse.(8)
The ophthalmologist can play a major role in diagnosis of this syndrome, because the ophthalmic phenotype could be sufficient to make the diagnosis of Knobloch syndrome, even in the absence of characteristic neurologic manifestations.(1) Regular ocular screening is necessary in these patients because of the risk of retinal detachment. We have described some OCT findings in KNO syndrome, which include a lack of the normal foveal pit, thinning of the neuroretina, subretinal scars, prominent choroidal vessels, and attenuation of the choriocapillaris and RPE. Our case shows that successful retinal detachment repair with a single surgery in a patient with KNO syndrome and relatively good final visual acuity (20/125) is achievable. | | | References | 1. Khan AO, Aldahmesh MA, Mohamed JY, Al-Mesfer S, Alkuraya FS. The distinct ophthalmic phenotype of Knobloch syndrome in children. Br J Ophthalmol 2012;96:890-5.
2. Hull S, Arno G, Ku CA, et al. Molecular and clinical findings in patients with Knobloch syndrome. JAMA Ophthalmol 2016;134:753-62.
3. Knobloch WH, Layer JM. Retinal detachment and encephalocele. J Pediatric Ophthalmol 1971;181-4.
4. Marneros AG, Keene DR, Hansen U, et al. Collagen XVIII/endostatin is essential for vision and retinal pigment epithelial function. EMBO J 2004;23:89-99.
5. AlBakri A, Ghazi NG, Khan AO. Biometry, optical coherence tomography, and further clinical observations in Knobloch syndrome. Ophthalmic Genet 2016;18:1-5. [Epub ahead of print]
6. Haghighi A, Tiwari A, Piri N, et al. Homozygosity mapping and whole exome sequencing reveal a novel homozygous COL18A1 mutation causing Knobloch syndrome. PloS One 2014;9:e112747.
7. Seaver LH, Joffe L, Spark RP, Smith BL, Hoyme HE. Congenital scalp defects and vitreoretinal degeneration: redefining the Knobloch syndrome. Am J Med Genet 1993;46:203-8.
8. Moysidis, SN, Aziz HA, Rachitskaya AV, Berrocal AM. Prophylactic scleral buckle implantation in Knobloch syndrome. J Pediatr Ophthalmol Strabismus 2014;51.e40-e43.
| |
|
|
|
|
|
|
 Welcome, please sign in
Welcome, please sign in