|
|
 |
|
|
|
|
|
Congenital hypertrophy of the retinal pigment epithelium complicated by a choroidal neovascular membrane
Digital Journal of Ophthalmology
2013
Volume 19, Number 2
May 1, 2013
DOI: 10.5693/djo.02.2013.01.004
|
Printer Friendly
Download PDF |
|
|
Pavlina Youhnovska, MD | Retina Service, Ophthalmology Department, CHUM Hôpital Notre-Dame, Montreal, Quebec, Canada Daniela Toffoli, MD | Emory Eye Center, Pediatric Ophthalmology Department, Atlanta Georgia Danny Gauthier, MD | Retina Service, Ophthalmology Department, CHUM Hôpital Notre-Dame, Montreal, Quebec, Canada
|
|
|
| Abstract | | A 66-year-old woman presenting with a history of visual loss in her left eye was diagnosed with congenital hypertrophy of the retinal pigment epithelium accompanied by a choroidal neovascular membrane (CNVM) affecting the macula. Visual acuity improved after 2 treatments of verteporfin photodynamic therapy. Recurrent CNVM in the same eye 3 years later was stabilized and vision slowly improved with a series of intravitreal bevacizumab injections. | | | Introduction | Congenital hypertrophy of the retinal pigment epithelium (CHRPE) is a benign clinical entity characterized by round, flat, dark gray or dark brown plaques, which are primarily found in the retinal mid-periphery but may also occur in more anterior locations or in the posterior pole, including the macula.(1,2) CHRPE lesions typically have well-defined, scalloped edges and contain central lacunae that appear “punched-out” in the retina. Additionally, they often have a hypopigmented “halo” lining the outer margins of the lesion. CHRPE typically presents as a solitary lesion; however, forms containing multiple small lesions have been reported.(3)
CHRPE typically shows little progression, but long-term follow-up studies have shown that the lesions occasionally grow slowly. Fluorescein angiography often reveals retinal vascular changes, including attenuation of the retinal capillary bed and the presence of microaneurysms.(2,4) | | | Case Report | A 66-year-old woman presented to CHUM Hôpital Notre-Dame, Montréal (PQ), Canada, in July 2005 with a 3-month history of decreased visual acuity and a central “dark spot” in her left eye. She denied other ocular symptoms, including metamorphopsia or micropsia. She had no other systemic complaints and was in good general health. On ophthalmological examination, visual acuity was 20/30 in the right eye and 20/80 in the left eye. Slit-lamp biomicroscopy was significant only for mild bilateral cataracts. Examination of the right fundus was unremarkable (Figure 1A). There were no drusen or retinal pigment epithelium (RPE) changes compatible with age-related macular degeneration (ARMD). Examination of the left fundus revealed a dark brown plaque, 1.5 disc diameters in size, confined to the superotemporal vascular arcade. The plaque was fragmented and contained central atrophic spaces and lacunae. Three similar but smaller lesions were observed adjacent to this plaque. These lesions had smooth, well-defined margins and were associated with atrophic changes. An area of extrafoveal thickening, surrounded by a ring of lipid exudates and two small hemorrhages within the foveal avascular zone were noted. These latter findings appeared compatible with the presence of a CNVM. No signs of ARMD were observed on left fundus examination (Figure 1B).
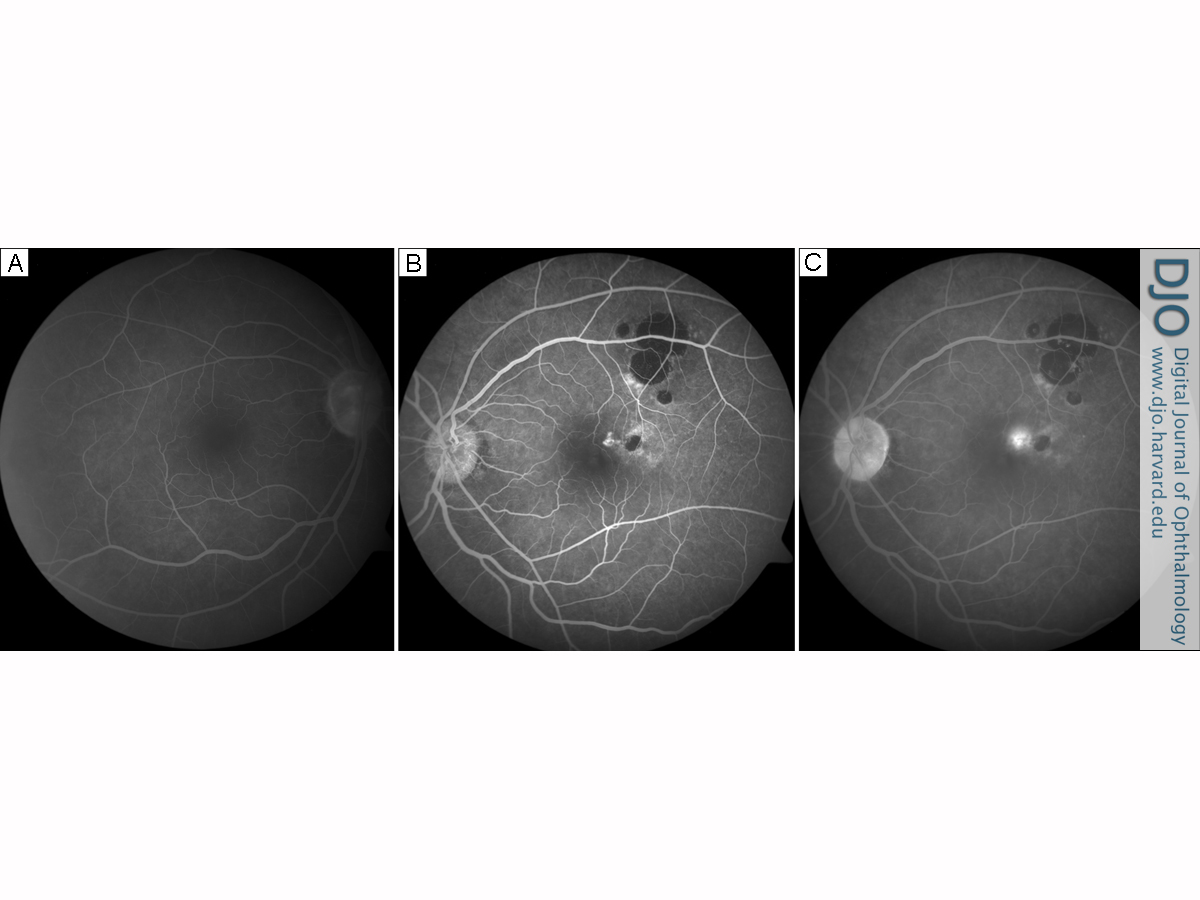
On fluorescein angiography, the right fundus was normal (Figure 2A). In the left eye, the pigmented lesions created a masking effect, corresponding to regions of hypofluorescence, whereas window defects were seen at the sites of atrophic changes. An area of extrafoveal hyperfluorescence was noted, arising immediately adjacent to one of the edges of a satellite lesion. Progressive capillary leakage was observed during the late phase of fluorescein transit, consistent with a classic CNVM (Figure 2B-C).
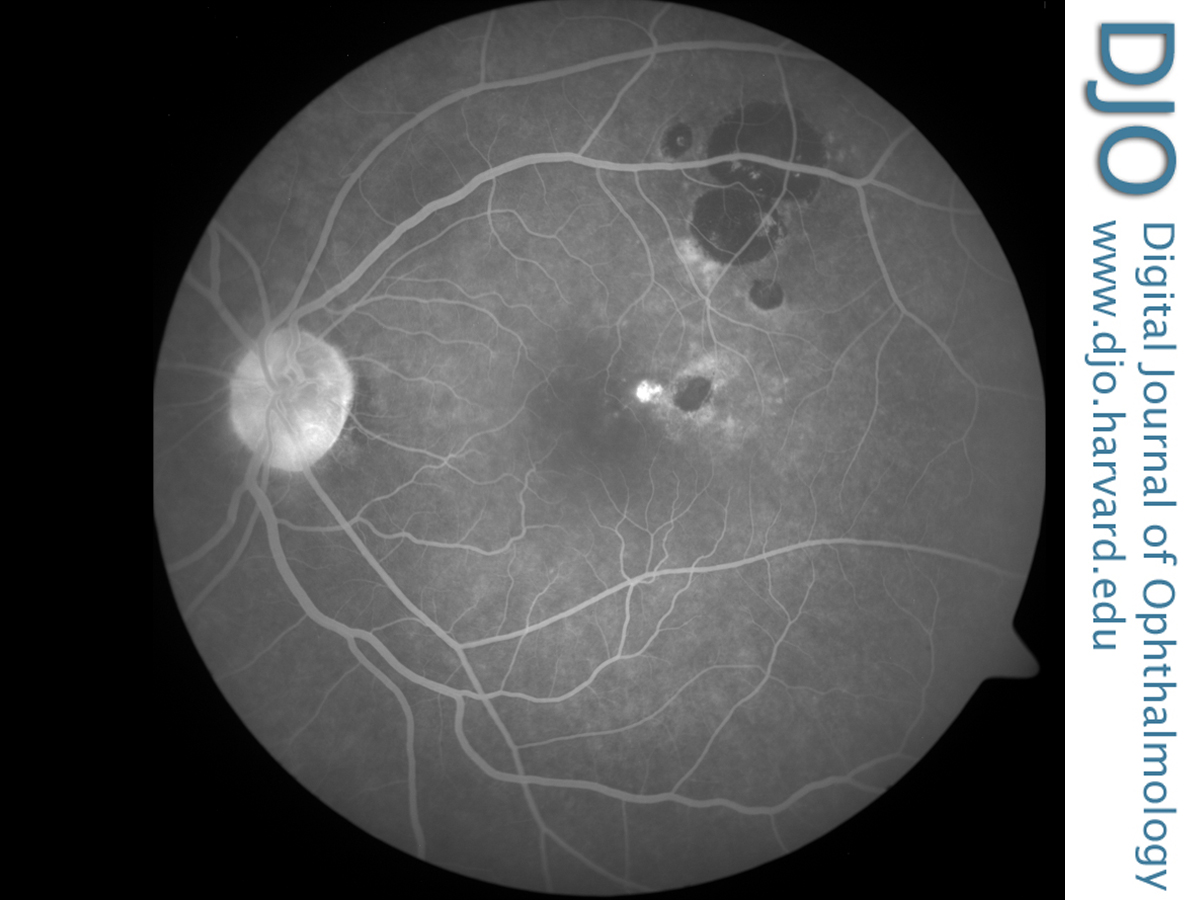
The patient was diagnosed with CHRPE complicated by a CNVM and treated with verteporfin photodynamic therapy (PDT). Standard parameters for fluence and irradiance were used for a total treatment time of 83 seconds. Following treatment, functional improvement was noted, with a visual acuity of 20/40 in the left eye at 6 months’ follow-up. There was no leakage of fluorescein noted on angiography after treatment, consistent with full regression of the CNVM (Figure 3). Clinical examination performed following two PDT treatments 3 months apart demonstrated a favorable response to the therapy with resolution of subretinal fluid and lipid exudates. The patient required no further treatment during the 2-year follow-up period.
Two years after completing treatment, she presented with a new episode of decreased vision in the same eye. On ophthalmological examination, visual acuity was 20/200 in the left eye. The fundus examination and the fluorescein angiography confirmed a recurrence of the macular CNVM. The patient was treated with intravitreal bevacizumab 1.25mg/0.05ml. Following 5 consecutive monthly injections, visual acuity stabilized at 20/60 and no CNV activity was noted at last follow-up, 2 years after her final injection. | |
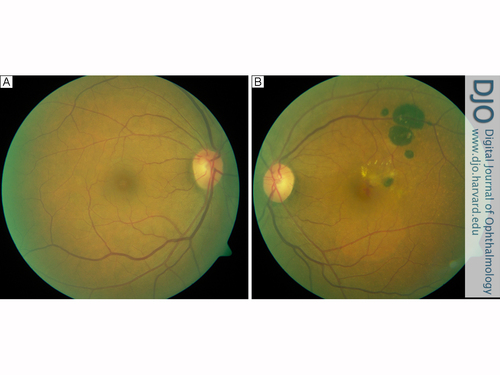
Figure 1
A, Color fundus photograph of the normal right eye. B, Color fundus photograph of the left eye showing a flat bi-fragmented pigmented dark brown plaque and 3 smaller, similar adjacent lesions consistent with congenital hypertrophy of the retinal pigment epithelium (CHRPE). Lacunae are present within the plaque and retinal thickening with a ring of lipid exudates and 2 small hemorrhages are seen in the macular area.
|
|

Figure 2
Fluorescein angiogram of the normal right eye at 6 minutes, 5 seconds. B,
Fluorescein angiogram of the left eye at 30.8 seconds. The CHRPE lesions show blockage hypofluorescence. The atrophy at the lesion’s margins gives rise to an RPE window defect. Early hyperfluorescence adjacent to the CHRPE plaque is seen in the juxtafoveal area. C, Fluorescein angiogram at 7 minutes, 6 seconds. The dye transit progresses to a late retinal leakage consistent with a choroidal neovascular membrane. The lacunae show an RPE window defect within the CHRPE hypofluorescent plaques.
|
|
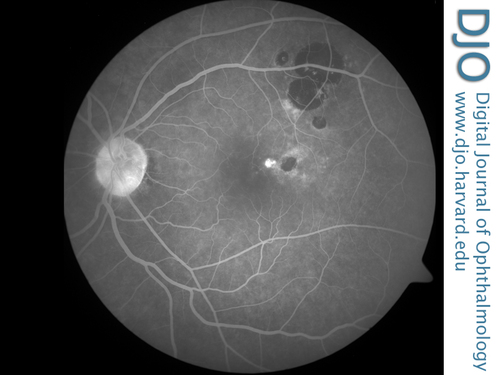
Figure 3
Late frame of the control fluorescein angiogram after verteporfin photodynamic therapy at 5 minutes, 46 seconds, showing an area of hyperfluorescence arising at the edge of the CHRPE lesion with absence of late leakage.
|
|
| Discussion | We describe a patient with macular CHRPE associated with a CNVM. The membrane was treated with verteporfin PDT and bevacizumab intravitreal injections. To our knowledge, there is only one previous case of CHRPE associated with neovascularization in the literature,(5) and this is the first report of CHRPE complicated by a CNVM in the macular area.
Retinal vascular changes, as documented by fluorescein angiography, may represent a sign of the CHRPE’s evolution in time. In a series of 12 patients, Cohen et al reported vascular abnormalities in 91% of 12 cases and suggested that the associated angiographic findings should be regarded as typical of the condition.(4) Vascular changes commonly observed include an attenuated capillary bed, microaneurysms, and chorioretinal anastomoses. It has been proposed that capillary abnormalities occur as a result of increased oxygen concentration in the inner retina. Abnormal fluorescein leakage has only been reported in 2 cases of CHRPE and in only 1 of the cases was fluorescein leakage noted at the margin of the lesion, increasing the suspicion of a CNVM.(5)
The precise etiology of the vascular changes noted in CHRPE have yet to be elucidated. Histological studies of CHRPE lesions demonstrate an abnormal RPE, with an increase in the height of individual cells and an increase in overall RPE cell density. Additionally, RPE cells in CHRPE lesions contain multiple granules and show an increase in pigment density compared to their normal counterparts. Photoreceptors overlying the abnormal RPE are also noted to undergo degeneration, a process that may lead to a relative or absolute scotoma. Although RPE cells adjacent to the CHRPE lesion may initially appear to be normal, they may also undergo change. Unlike the increase in height of the RPE cells seen within the CHRPE plaque, however, they tend to broaden and flatten out. Some authors believe that these flatter RPE cells may induce pressure on the hypertrophic RPE cells present within the CHRPE lesion, creating areas of micro-trauma at the junction between the two.(2) Breaks in the RPE associated with these areas of micro-trauma may be associated with RPE cell migration into the inner retina and may allow the subretinal fibrovascular growth associated with neovascular membranes. This may provide a possible explanation for CNV proliferation found at the junction of CHRPE lesions and adjacent normal tissue.
In our case, the lesion was fragmented and associated with three satellite lesions, which may have resulted from breaks in the RPE and possibly Bruch’s membrane and subsequent migration of hypertrophic epithelial cells. This contrasts with the behavior of a typical CHRPE plaque, where migration of RPE cells results in enlargement of atrophic lacunae with subsequent concentric enlargement of the lesion. Other typical progressive CHRPE lesions include plaques with poorly pigmented linear streaks. Chamot et al suggested that the same mechanism could be a possible explanation for the natural history of both forms.(2)
When CHRPE presents with multiple lesions, it must be differentiated from the multifocal epithelial hypertrophy associated with Gardner’s syndrome. The latter pathology presents with multiple, bilateral, variably pigmented retinal lesions and is associated with familial adenoidal polyposis. Any suspicion of Gardner’s syndrome requires further systemic work-up, including colonoscopy. Also to be included in the differential diagnosis of CHRPE is the congenital grouped hypertrophy of the RPE. This pathology typically presents with numerous pigmented retinal spots resembling “bear tracks” and is not associated with vascular abnormalities on fluorescein angiography. In some cases, it can be found alongside a typical solitary CHRPE.(4) Finally, the angiographic and ultrasound characteristics of a choroidal melanoma make it easy to differentiate from solitary forms of CHRPE.
CHRPE is a clinical diagnosis. The lesion is usually asymptomatic and is found incidentally on routine ophthalmoscopic examination. Progressive changes are now considered to be a part of the natural history of CHRPE; the evolution is not a sign of malignancy since the lesion remains benign. Fluorescein angiography should be performed to reveal the presence of associated retinal vascular abnormalities, especially if a CNVM is suspected. CNVMs, particularly in the macular area, are an extremely rare complication of CHRPE. However, as in our case, CNV can be a vision-threatening condition and should be identified and managed rapidly. | | | References | 1. Nishikatsu, H, Shiono, T. Congenital hypertrophy of the retinal pigment epithelium in the macula. Ophthalmologica 1996; 210:126-8.
2. Chamot, L, Zografos, L, Klainguti, G. Fundus changes associated with congenital hypertrophy of the retinal pigment epithelium. Am J Ophthalmol 1993;115:154-61.
3. Buettner, H. Congenital hypertrophy of the retinal pigment epithelium. Am J Ophthalmol 1975; 79:177-89.
4. Cohen, SY, Quentel, G, Guiberteau, B, Coscas, GJ. Retinal vascular changes in congenital hypertrophy of the retinal pigment epithelium. Ophthalmology 1993;100:471-4.
5. Cleary, PE, Gregor, Z, Bird, AC. Retinal vascular changes in congenital hypertrophy of the retinal pigment epithelium. Br J Ophthalmol 1976; 60:499-503.
| |
|
|
|
|
|
|
 Welcome, please sign in
Welcome, please sign in