|
|
 |
|
|
|
|
|
Retinoblastoma
|
Printer Friendly
|
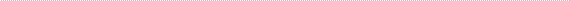
Peter K. Kaiser, MD Ingrid U. Scott, MD Joan M. O'Brien, MD Timothy G. Murray, MD Bascom Palmer Eye Institute, University of Miami School of Medicine January 13, 2003
|
What is Retinoblastoma?
Although the disease is very rare, retinoblastoma (RB) is the most common eye tumor in children, and the third most common cancer overall affecting children. Retinoblastoma is a disease that causes the growth of malignant tumors in the retinal cell layer of the eye. The frequency that retinoblastoma occurs has increased over the past 60 years. It now occurs in 1 out of every 15,000 live biths. Two hundred fifty to 350 new cases are diagnosed each year in the US with over 90 percent of cases presenting before the age of 5 years. Untreated, retinoblastoma is almost always fatal; therefore, early diagnosis and treatment is critical in saving lives and preserving visual function.
The treatment of retinoblastoma depends upon whether or not one or both eyes are involved and the extent of tumor. Although removing the eye (enucleation) remains a frequent treatment for retinoblastoma, conservative strategies are being increasingly employed. With earlier detection and improved treatment modalities, the prognosis for vision and life in patients with retinoblastoma has improved significantly in the last twenty years.
How Can I Tell If My Child Has Retinoblastoma?
Most pediatricians routinely screen children for eye abnormalities during well-baby examinations. However, it is important for parents to evaluate their children between these screenings for potentially serious eye disease. The diagnosis of retinoblastoma is usually made after signs of the disease are noted by the parents or pediatricians. A few patients may be examined in early infancy because of a family history of retinoblastoma; however, only about five to eight percent of patients have a positive family history. Moreover, although all babies are screened at birth for eye problems, retinoblastoma is not necessarily present at this time. It may develop later; thus, it is important to have your child examined regularly. Finally, these tests do not eliminate the need for well-baby visits with your primary care provider or pediatrician.
If you suspect that these abnormalities are present and not adequately explained by your pediatrician, you should seek an eye examination by an ophthalmologist specializing in pediatric eye disease (ophthalmologist). If the ophthalmologist detects abnormalities, an examination under anesthesia may be required to further evaluate your child.
- The most common presenting sign of retinoblastoma is seeing a whitish color behind the pupil which is usually black (leukocoria). This sensitive test is best done by looking at the "red reflex." Most parents are familiar with the red reflection one often gets in children's eyes when taking flash photographs. The red reflex can also be elicited by dimming the room lights and using a flashlight to shine light directly INTO the child's eyes. When leukocoria, also known as "cat's eye" is present the red reflex is absent or incomplete. This abnormality is present in approximately 60% of all children with retinoblastoma.
- The second most common finding in retinoblastoma is strabismus or problems with eye movements (crossed eyes). Parents can test their children by evaluating eye symmetry in normal lighting. The eyes should appear normal and equal in size. They should move together and focus normally. The two eyes should be aligned normally and should not wander or cross. Another way to test symmetry is to cover your child's eyes separately with your hand. First cover one eye, then move your hand over the other eye. The eyes should remain aligned and not move in or out when doing this test. Twenty percent of patients with retinoblastoma will manifest strabismus.
- If your child develops a red irritation that persists, it may represent inflammation or pseudo-inflammation in the eye. Ten percent of patients with retinoblastoma can develop this symptom.
- Other rare presenting signs of retinoblastoma include differences in pupil size (anisocoria), differences in iris color (heterochromia), tearing, bulging forward of the eyes (proptosis), cataract, and abnormal eye movements (nystagmus).
Who Gets Retinoblasoma?
Retinoblastoma is a childhood disease. In general, most new cases of retinoblastoma are diagnosed before the age of 5. Children with retinoblastoma in both eyes are generally diagnosed at a younger age (mean = 13 to 15 months) than children with unilateral disease (mean = 24 months). The overall average age at diagnosis is 18 months. There is no known sex or racial predilection. Approximately 60 percent of cases are only in one eye when first diagnosed, unfortunately about 40 percent of cases present in both eyes.
Retinoblastoma can be either hereditary or non-hereditary. It is hereditary in 30 to 40 percent of patients. Thus, all children in a family may be at risk. Overall, only six percent of newly diagnosed retinoblastoma patients have a positive family history for the disease. Of the patients with hereditary disease, 25 percent have a positive family history. The remaining 75 percent of hereditary cases are caused by new mutations or by inheritance FROM a parent who carries the gene, but does not have any symptoms. Retinoblastoma does not develop in approximately ten percent of patients who carry the heritable mutation.
Hereditary retinoblastomas usually occur at a younger age and are more likely bilateral and multicentric. In addition, the patient has a higher risk of developing tumors in other parts of the body. It has been reported that patients with bilateral retinoblastoma have approximately a 5 percent chance of developing another cancer during the first 10 years of follow-up, 18 percent within the first 20 years, and 26 percent within the first 30 years. The average time between presentation with retinoblastoma and the appearance of a second cancer is between 10.4 and 13 years. The most common non-ocular cancer is osteogenic sarcoma, other tumors such as rhabdomyosarcoma, chondrosarcoma, spindle cell sarcoma, neuroblastoma, glioma, leukemia, sebaceous cell carcinoma, squamous cell carcinoma, and malignant melanoma have also been reported.
What are the Chances of Other Children HAVING RB?
Genetic counseling is an important aspect in the treatment of retinoblastoma. The key is identifying patients who carry the heritable mutation, since these patients have an approximately 80 to 90 percent chance of developing retinoblastoma and a 50 percent chance of passing the gene to their future children. Several clinical features help to identify children that may carry the heritable form of retinoblastoma. Patients with bilateral or multifocal retinoblastomas and those with a positive family history of retinoblastoma are assumed to have the heritable retinoblastoma mutation. It is important to remember that the retinoblastoma gene produces cancer in approximately 80 to 90 percent of patients who carry the gene and, therefore, about 40 percent of the patients' offsprings will have retinoblastoma, while some offspring may simply carry the gene and never develop the disease.
Because of the hereditary nature of retinoblastoma, it is currently recommended that all siblings and children of retinoblastoma patients be examined under anesthesia (EUA) every 2 to 4 months during the first years of life. In the future, laboratory studies including karyotyping, Southern blot analysis, Polymerase Chain Reactions (PCR), DNA sequence analysis/polymorphisms, and Esterase D reactions may eliminate the need to examine children under anesthesia. The aim of genetic screening in families with hereditary retinoblastoma is to identify those persons who carry a mutation and are at risk for tumor development. In 15 percent of families with hereditary retinoblastoma, the tumor-predisposing mutation itself can be detected using karyotyping or Southern blot analysis. When a detectable deletion is present, one can identify individuals carrying the mutation with over 99 percent accuracy. In the remaining 85 percent of families, no mutation can be found directly. However, in 95 percent of such families, DNA sequence polymorphisms allow the identification of the copy of the gene that carries the tumor-predisposing gene. Predictions based on this analysis are over 95% accurate.
How Do You Treat Retinoblastoma?
- Enucleation (removal of the eye)
- External beam radiation therapy (radiation treatment)
- Localized plaque radiation therapy (radiation treatment)
- Photocoagulation (laser treatment)
- Cryotherapy (freezing treatment)
- Chemotherapy
Note: Some of these treatment expanations are very detailed and medically oriented for health care professionals. These descriptions are only guidelines of currently accepted treatment protocols. Every case of retinoblastoma is unique, and the treatments, or combination of treatments, vary according to size, shape, and location of the tumor, whether both eyes are affected or not, and whether or not the tumor has spread (metastasis). Patients and parents should consult their physicians for specific treatment options.
Enucleation (removal of the eye)
Because of earlier tumor detection, as well as improved and increased use of more conservative eye-sparing treatments, there has been a significant decrease in the frequency of enucleation in patients with retinoblastoma over the past forty years. Unfortunately, however, enucleation remains a frequent treatment for retinoblastoma, and is indicated for all unilateral tumors that fill over half of the eye, or when there is extensive seeding of tumor INTO the jelly that fills the eye (vitreous), total detachment of the retina (the back of the eye), new blood vessel growth on the iris, or involvement of other eye structures by the tumor. Historically, in patients with bilateral retinoblastoma, the eye with more advanced tumor has been enucleated and the less involved eye managed by other methods. Very rarely, if the disease is far advanced in both eyes, bilateral enucleation is justified.
When a child has their eye removed to treat retinoblastoma, during surgery an adult size implant is placed INTO the orbit to allow the eye muscles to be reattached. This allows more natural eye movements in the future. The implant is usually made FROM hydroxylapatite, a derivative of coral, this allows blood vessels to grow INTO the implant and seal it INTO place. Shortly after surgery, a shell-like prosthesis is fitted over the implant. The prothesis is made to match the color and shape of the other eye giving the child a natural appearance.
External Beam Radiotherapy (Radiation Treatment)
Because retinoblastoma is extremely sensitive to radiation, irradiation can be an effective treatment. External beam radiotherapy is most often used to treat patients with bilateral retinoblastoma who are not amenable to local treatment (see below). This method of treatment is generally preferred when tumor recurs or extends INTO the bones around the eye (orbit), when the second eye contains a tumor larger than 16 mm in diameter, when the tumor is near the optic disc or center of vision (fovea), when multiple tumors are present, or when there is extensive vitreous seeding. It may also be used to treat the eye socket after an eye has been removed if studies SHOW extension beyond the area that had been removed.
The currently accepted protocol is 3,500 to 4,000 centigrays (cGy) of external beam irradiation delivered in divided doses over 4 to 5 weeks. Complications FROM radiation treatment include cataract, radiation retinopathy, optic neuropathy, dry eye, sunken orbit, atrophy of muscle and subcutaneous tissues, and periorbital bone maldevelopment (mid-facial hypoplasia). An important long-term complication of external beam radiation is the development of radiation-induced tumors. Several studies suggest that patients with hereditary retinoblastoma have an increased incidence of secondary tumors, and that the incidence rate is further increased in patients who receive radiation therapy.
Plaque Radiotherapy (Radiation Treatment)
Plaque radiotherapy (local treatment) has advantages over external beam radiotherapy in that it delivers radiation in a more localized fashion, thereby minimizing exposure to other eye structures. Theoretically, then, plaque radiotherapy should minimize such complications as the development of radiation-induced secondary tumors. However, plaque radiotherapy may be associated with higher rates of radiation-induced retina problems (retinopathy) and optic nerve problems (papillopathy) than external beam radiation. Plaque radiotherapy also has advantages over cryotherapy and photocoagulation in that the former can be effective for tumors up to 16 mm in diameter and 8 mm in thickness. In contrast, cryotherapy and photocoagulation are generally ineffective treatment modalities for tumors greater than 6 mm in diameter and/or 3 mm in thickness.
Plaque radiotherapy for retinoblastoma generally consists of 3,500 to 4,000 cGy to the tumor apex, and most tumors respond dramatically within the first 3 weeks. It should be stressed that only a small percentage of patients with retinoblastoma are amenable to treatment with plaque radiotherapy. In general, this treatment modality may be used in patients with small, solitary tumors (up to 16 mm in diameter and 8 mm in thickness).
Photocoagulation (Laser Treatment)
Photocoagulation can be used to treat selected small tumors that do not involve the optic disc or macula. In tumors less than or equal to 3.0 mm in diameter and 2.0 mm in thickness, and confined to the retina without vitreous seeding, photocoagulation was generally successful. The success of laser treatment depends on several factors including tumor size (97.2 percent of tumors up to 1 disc diameter in size were cured, as compared to 40.9 percent of tumors larger than 5 disc diameters), tumor location (82.7 percent of tumors anterior to the equator were treated successfully as compared to 59.5 percent of tumors posterior to the equator), and tumor elevation (80.6 percent of tumors with height equal or less than half the basal diameter were successfully treated as compared with 43.4 percent of tumors with height larger than half the basal diameter).
The recommended technique for treating retinoblastomas with photocoagulation consists of placing 1 or 2 rows of confluent burns around the tumor and, where appropriate, over the tumor surface, using sufficient power to produce whitening of the retina and closure of the retinal vessels that supply the tumor. The tumor should regress within several weeks although multiple photocoagulation sessions may be necessary. With proper administration, photocoagulation for retinoblastoma is associated with few complications.
Cryotherapy (Freezing Treatment)
Cryotherapy may be used as a primary or secondary treatment of peripheral retinoblastomas. Unlike photocoagulation, cryotherapy may be effective for vitreous seeding less than 0.5 mm FROM the tumor apex. In general, cryotherapy is effective for tumors up to 5.0 mm in diameter and 3.0 mm in thickness, although multiple treatment sessions may be necessary.
Cryotherapy for retinoblastoma should be administered by the triple freeze-thaw technique. The treatment may need to be repeated, typically at 3 to 4 week intervals, if viable tumor is evident on clinical examination. A successfully treated retinoblastoma appears as a flat, well-delineated variably pigmented scar with no signs of viable tumor. Local vitreous hemorrhage and transient subretinal fluid have been reported complications of cryotherapy for retinoblastoma.
Chemotherapy
There has been increasing interest in using systemic chemotherapy as adjuvant therapy in children previously considered candidates only for enucleation or bilateral external beam radiotherapy. It is believed that using chemotherapy for tumor reduction may make tumors amenable to focal treatment or permit treatment with a lower dosage of radiation. This, in turn, may decrease the incidence of secondary tumors and minimize such complications as mid-facial hypoplasia. In addition, there may be a role for pre-enucleation chemotherapeutic induction in children with high-risk features such as orbital disease or suspected optic nerve disease.
The observed short-term adverse effects of adjuvant chemotherapy are well-tolerated. Like radiotherapy, chemotherapy (particularly alkylating agents) has been reported to increase the incidence of second tumors. In addition, alkylating agents may be associated with the development of nonlymphoblastic leukemia and infertility.
What is the Prognosis?
Among patients with unilateral disease, the visual prognosis for the uninvolved eye is generally excellent. Among patients with bilateral disease, the visual prognosis depends on the location and extent of involvement as well as treatment efficacy. One study reported that among patients with bilateral disease treated conservatively, 50 percent achieved vision of 20/40 or better in one eye by the age of 8 years.
The increasing survival of patients with retinoblastoma has been attributed to earlier diagnosis and improved methods of treatment. The death rate FROM retinoblastoma was nearly 100 percent a century ago, compared with 18 percent in 1964 and less than 10 percent in 1990. It has been reported that survival is greatest among patients diagnosed before 2 years of age or after 7 years of age. As our ability to treat and diagnose retinoblastoma improves, we can only hope that the survival rate will continue to improve.
Where Can I Get More Information?
The National Retinoblastoma Research & Support Foundation | |
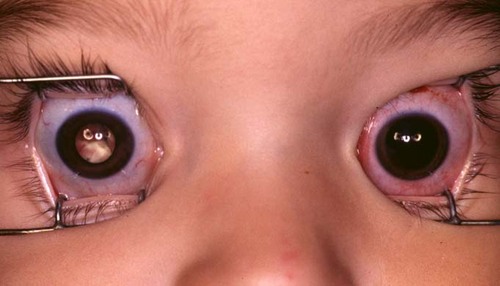 Figure 1
Figure 1
|
|
 The National Retinoblastoma Research & Support Foundation
The National Retinoblastoma Research & Support Foundation
|
|
|
The information and recommendations appearing on these pages
are informational only and is not intended to be a basis for diagnosis, treatment
or any other clinical application. For specific information concerning your personal
medical condition, the DJO suggests that you consult your physician.
|
|
|
|
|
|
|
 Welcome, please sign in
Welcome, please sign in 


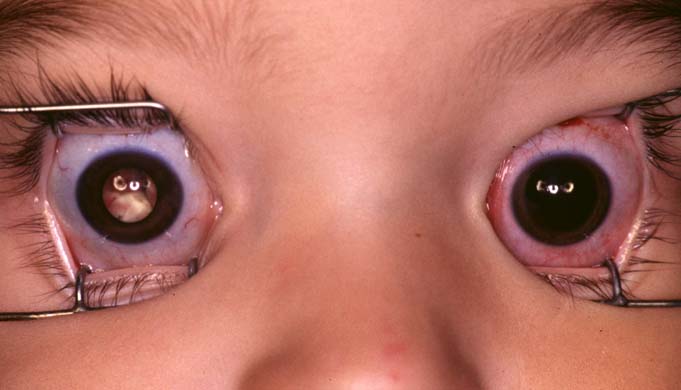 Figure 1
Figure 1 The National Retinoblastoma Research & Support Foundation
The National Retinoblastoma Research & Support Foundation