|
|
 |
|
|
|
|
|
A 59 year old woman with acute visual field loss
Digital Journal of Ophthalmology 2004
Volume 10, Number 9
September 15, 2004
|
Printer Friendly
|
|
|
|
|
|
|
| Diagnosis and Discussion | The most likely provisional clinical diagnosis prior to scanning was suprasellar infrachiasmatic lesion such as pituitary adenoma causing compression of the chiasm from below and giving rise to asymmetrical bitemporal superior quadrantanopia (chiasmal syndrome). The CT revealed typical findings of craniopharyngioma. This diagnosis was later confirmed by histopathology (figure 7).
Craniopharyngioma is rare, and accounts for 1-3% of intracranial tumours (5-10% in children) and 13% of suprasellar tumours (56% in children). There is slight male preponderance and no genetic relationship has been found. There is a bimodal peak, the largest peak being at the age of 5-14 years and a much smaller second peak at 50-70 years of age. The overall global incidence is 0.5-2 per 100,000 per year (1).
Craniopharyngioma is a slow-growing, ectodermal tumour arising from squamous cell rests derived from primitive buccal epithelium (Rathke's pouch) and occurring mainly on the infundibulum between the inferior surface of the brain and superior surface of the pituitary . Embryogenetic and metaplastic theories have been put forward to explain bimodal peak (2).
Macroscopically the tumour my be cystic, solid or, often, a mixture, and there is often focal calcification (produced by chronic reaction to leaking cyst contents). The cysts contain glittering, turbid, brownish-yellow fluid that is said to resemble machine oil.
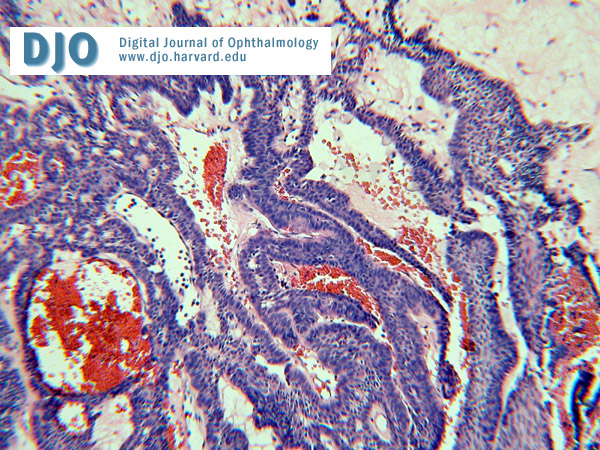
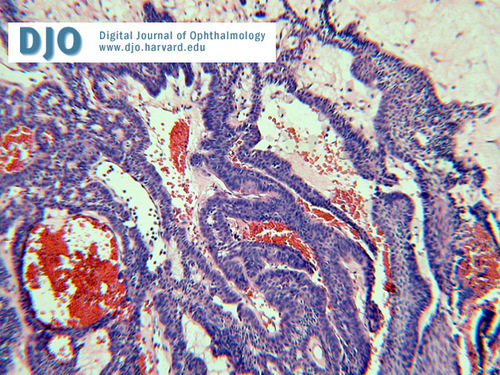
Microscopically it is composed of broad strands and cords of a multi- stratified squamous epithelium, most commonly of the adamantinomatous type with peripheral palisading as seen in figure 7. Compact nodules of "wet" keratin and dystrophic calcification are commonly seen. The papillary variant lacks the palisading and wet keratin formation.
Initial clinical manifestations of this tumour depend on the precise site of origin, the direction of the growth and the age of the patient. Three major clinical syndromes have been described relating to the anatomic relation of the craniopharyngioma with surrounding structures. Prechiasmal localization typically results in visual disturbances (37-68%). Retrochiasmal extension affects hypothalamus and commonly results in hydrocephalus and mental status abnormality (37-68%). Intrasellar craniopharyngioma usually manifests with headache (55-86%) and endocrinopathy (66-90%). Children are remarkably resistant to visual failure (2,3).
Ophthalmologists are often the first port of consultation and patients may present with headache, unilateral or bilateral visual loss, visual field loss, hemislide phenomenon, post fixational blindness, see-saw nystagmus, cranial nerve palsy, papilledema, and bow tie optic atrophy. Besides causing the chiasmal syndrome most commonly, craniopharyngioma has infrequently been reported to cause junctional scotoma, paracentral bitemporal hemianopia(anterior or posterior optic chiasma syndrome) or homonymous hemianopia (optic tract syndrome). However because of the suprachiasmatic location of the tumour there is a clear association between inferior visual field defects and craniopharyngioma, but in clinical practice most patients do not present with inferior field loss (as in our patient) (4). Pleomorphism- distinct change from one type of field defect into another type of visual field defect- is characteristic of craniopharyngioma. It is related to intermittent emptying of cyst fluid into ventricular system, a feature also responsible for fluctuating visual symptoms and masquerading acute disease processes like optic neuritis, infection or rapidly enlarging aneurysm (5).
Diagnostic studies include neuroimaging, endocrinologic and neuropsychologic work up. CT scan is the most sensitive and defines both calcified and cystic parts. MRI is important to plan surgical approach. Inspite the history of cold intolerance and weight gain, all endocrine workup in our patient was normal.
Craniopharyngiomas are histologically benign, but exhibit clinically malignant behaviour because of their proximity to important intracranial structures and their tendency to recur after what was thought to be total resection. There is yet no consensus on various treatment modalities. The options are total resection or subtotal resection with radiotherapy supported by hormonal replacement therapy. Other less tried treatment options are intracystic bleomycin, external beam radiation, and sterotactic radiation. Our patient underwent craniotomy and gross total resection with radiotherapy. Six months following her surgery her bilteral visual acquities were 6/5 and her postoperative fields had significantly improved. (figures 8 and 9).
| |
|
Figure 7
Micrograph of the pathology specimen.
 |
|
|
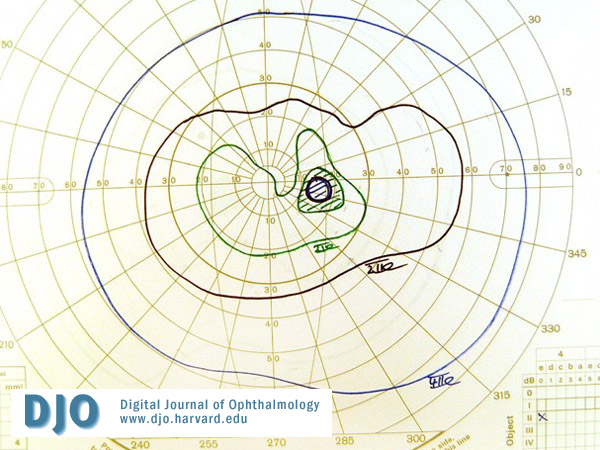
Figure 8
Post-operative visual field OS.
 |
|
|
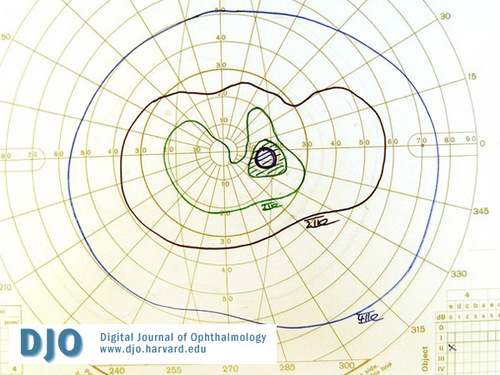
Figure 9
Post-operative visual field OD.
 |
|
|
|
|
|
|
|
 Welcome, please sign in
Welcome, please sign in