|
|
 |
|
|
|
|
|
27 year old woman with blurred central vision OS and headache
Digital Journal of Ophthalmology 1997
Volume 3, Number 18
May 15, 1997
|
Printer Friendly
|
|
|
|
|
|
|
| Diagnosis and Discussion | Vogt-Koyanagi-Harada's Disease (Uveomenigitic Syndrome)
Background:
Vogt in 1906 and Koyanagi in 1929 both described patients with bilateral anterior uveitis, vitiligo, poliosis, alopecia, and dysacusia. Harada in 1926 described patients with posterior uveitis, exudative retinal detachment, and cerebrospinal fluid pleocytosis. Over time it was recognized that there was significant overlap in the clinical characteristics of these patients and that these different clinical features were manifestations of the same disease. Hence, ophthalmologists began to call this constellation of clinical findings Vogt-Koyanagi-Harada syndrome or simply VKH. Some authors have also called this condition uveo-meningitic syndrome because of the frequent combination of uveitis and meningitis.
Epidemiology:
This condition has an equal gender distribution and usualy occurs between the ages of 20 to 50. VKH may occur in persons of any ethnic group, but is more common in Asians, Native Americans, and more darkly pigmented races. VKH is particularly common in Japan, where it represents 8% of all uveitis cases there. There is also an an association between VKH and certain HLA types including HLA-Bw54, HLA-DR4, and HLA-DqWa.
Clinical Features:
Although VKH is believed to be one single entitiy, it is clinically useful to differentiate between the Vogt-Koyanagi variant and the Harada's variant. The following TABLE points out some of the more important differences between the different possible presentations of VKH.
"0">
|
|
Vogt-Koyanagi Syndrome |
Harada's Disease |
Ocular Symptoms |
severe granulomatous anterior uveitis |
vitritis, choroiditis, exudative retinal detachment common |
CNS Symptoms |
mild or lacking |
often severe, including headache, encephalitis, cranial nerve palsies
and psychosis |
Ocular Sequelae |
cataracts, posterior synechiae, glaucoma |
hypopigmentation of the fundus after reattachment of retina producing
a "sunset glow" |
Poliosis or Alopecia |
90% |
<10% |
Vitiligo |
>50% |
<10% |
Auditory Disturbances |
common (>50%) |
less common |
Visual Prognosis |
poor |
fair to good |
Table 1
Pathophysiology:
VKH is believed to be an auto-immune disorder. There are increased levels of anti-retinal antibodies in patients with VKH. Lymphocytes have been shown to be sensitized to myelin basic protein in this condition, which may help to EXPLAIN some of the central nervous system findings. As noted before there are also an HLA associations in this condition.
Histopathology:
Classically, VKH is described as a granulomatous inflammation, but it may also be non-granulomatous. VKH appeas similar to sympathetic ophthalmia pathologically, but differs in that the choriocapillaris and retina are involved in VKH, but are typically spared in sympathetic ophthalmia.
Diagnosis:
The Vogt-Koyanagi variant is usually diagnosed based on characteristic examination findings e.g. granulomatous anterior uveitis associated with poliosis and vitiligo. Several studies are helpful in the diagnosis of the Harada's variant. Fluorescein angiography usually shows pinpoint areas of hyperfluorescence at the level of the retinal pigment epithelium early in the study, followed by late leakage. Ultrasound demonstrates thickening of the choroid in Harada's disease and is helpful to rule out other causes of exudative detachment such as posterior scleritis or tumor. A spinal tap may SHOW cerebrospinal fluid pleocytosis in Harada's disease, especially with meningeal involvement.
Treatment:
First line treatment of VKH is steroids (e.g. Prednisone 1.5 mg/Kg/day). If steroids are not sufficient, immunosuppressive agents such as cyclosporine and chlorambucil may be necessary.
Our patient was treated with 60 mg of prednisone P.O. qd. | |
|
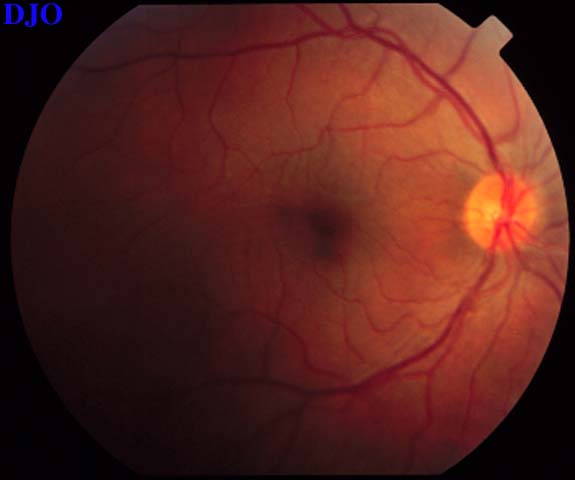
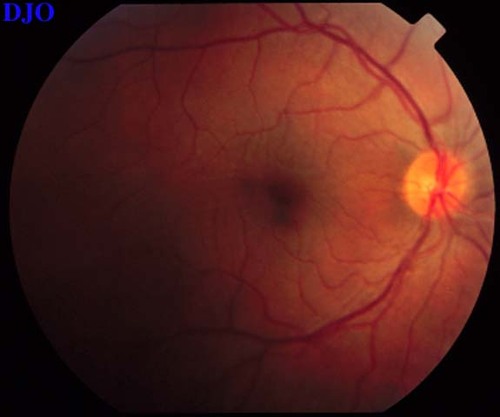
Figure 9
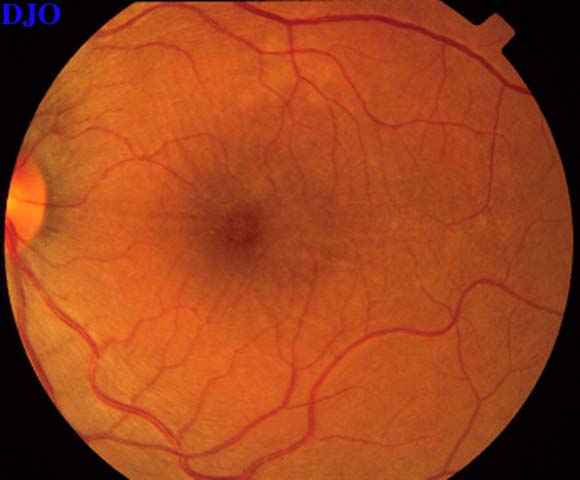
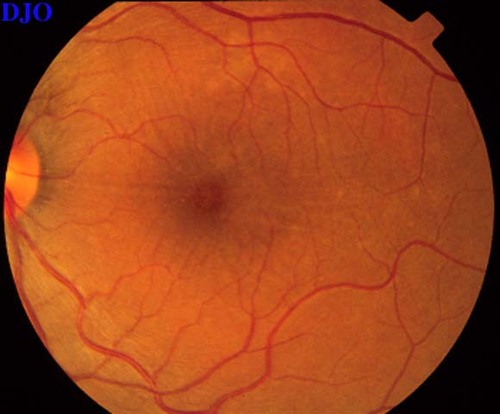
Figures 9-10. On the left the patient's fundus OS is shown which demonstrates the serous macular detachment. On the right, we see the patient's fundus OS after 1 week of the oral steroids. The serous detachment has largely resolved, but there remain striae in the macula. The patient's vision had improved FROM 16/200 to 20/50 OS.
 |
|
|
Figure 10
 |
|
|
|
 Welcome, please sign in
Welcome, please sign in