A 72-year-old man with double vision
Digital Journal of Ophthalmology 2007
Volume 13, Number 7
February 17, 2007
Volume 13, Number 7
February 17, 2007
Sreekumari Pushpoth | Wolverhampton and Midlands Eye Infirmary
Stephanie Smith | East Lancashire Hospitals NHS Trust
J.P. Roper | East Lancashire Hospitals NHS Trust
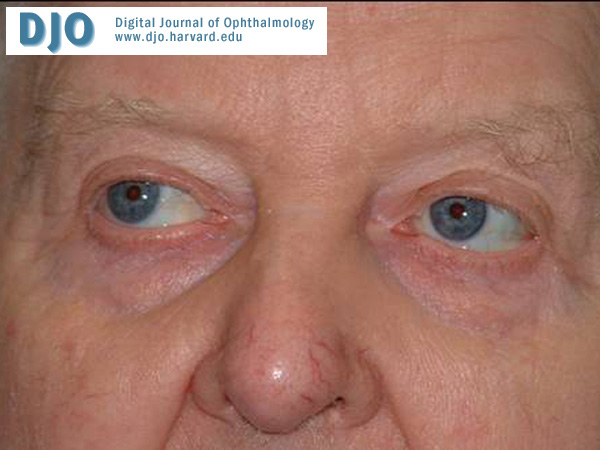
Figure 1
Attempted upgaze
Attempted upgaze
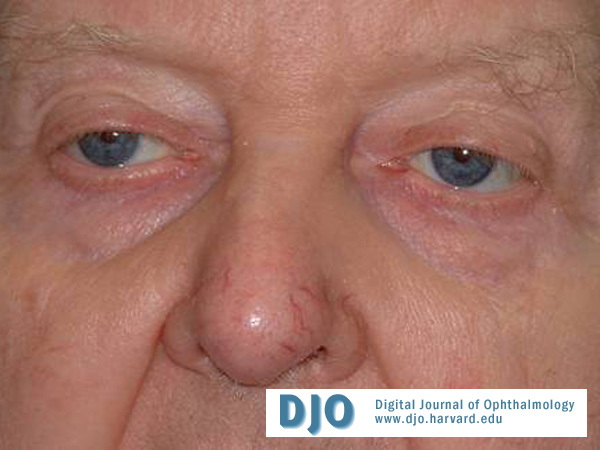
Figure 2
Attempted down gaze
Attempted down gaze
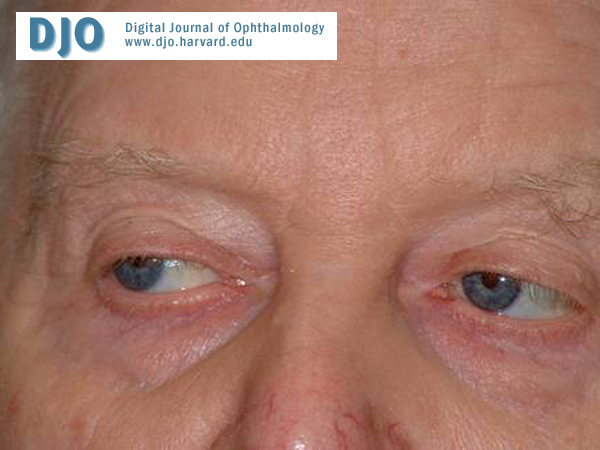
Figure 3
Right gaze
Right gaze
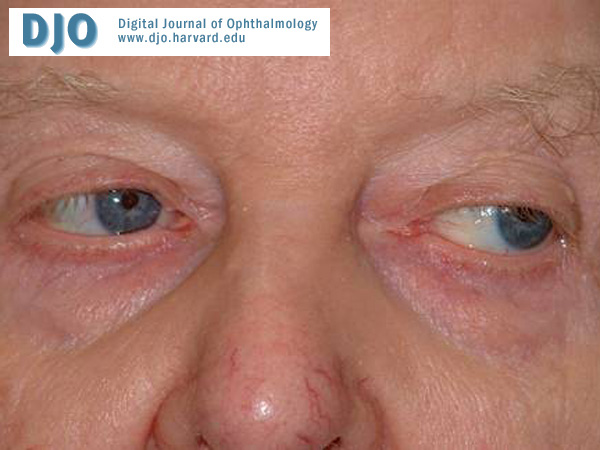
Figure 4
Left gaze
Left gaze
CT scan showed generalized cortical atrophy.
Whipple’s Disease
Diffuse Lewy body disease
Myasthenia Gravis
Thyroid eye disease
Progressive supranuclear palsy (PSP), (also known as Nuchal Dystonia Dementia Syndrome, Steele-Richardson-Olszewski Syndrome) is an important, often under-diagnosed neurodegenerative syndrome, which affects the brainstem and basal ganglia. It was first described as a distinct syndrome by John Steele, J Clifford Richardson and Jerzy Olszewski in 1963.
Patients usually present with postural instability with frequent falls. In the earliest stages of the disease there may be slowness of vertical saccadic eye movements which progress to limitation of downwards vertical saccadic eye movements and then to a complete vertical gaze palsy.(1) The doll's head maneuver demonstrates a normal vertical vestibular-ocular response. This confirms the integrity of the third nerve nuclei and places the pathology of the eye movement at supranuclear level.(2) Over activity of the frontalis muscle combined with diminished blink rate lead to a “surprised” facial appearance. Eye opening may be impaired either by active involuntary contraction of orbicularis oculi (blepharospasm) or by inability to voluntarily open the eyes (“apraxia of eyelid opening”).(3) Patients may also have cognitive dysfunction, axial rigidity, dysphagia and dysarthria.
Patients usually present with symptoms between 50 and 77 years of age.
The disease process is persistently progressive. Supportive treatment is the mainstay of management. Physiotherapy and occupational therapy are important in helping with aids for balance and avoidance of falls. Another difficulty faced by these patients is with swallowing. Speech therapy services have an important role in swallowing assessment, mainly to avoid the complications of aspiration.(4)
The prevalence of PSP is often underestimated as it is often misdiagnosed as Parkinson’s Disease (PD).(5, 6) It is very important to differentiate between the two, as patients with PSP need more supportive therapy. Features like taut spastic face, dysarthria, neck held in extension with axial rigidity, a symmetrical mild distal bradykinesia, normal muscle tone and absence of rest tremor helps to clinically differentiate PSP from PD.
Better understanding of the disease process and its pathology may help in the identification of newer disease modifying agents for these patients.
2. Huw R Morris, Nicholas W Wood, Andrew J Lees Progressive supranuclear palsy (Steele-Richardson-Olszewski disease) Postgrad Med J 1999;75:579-584.
3. Golbe LI, Davis PH, Lepore FE. Eyelid movement abnormalities in progressive supranuclear palsy. Mov Disord 1989; 4:297-302.
4. Litvan I, Sastry N, Sonies BC. Characterizing swallowing abnormalities in progressive supranuclear palsy. Neurology 1997;48:1654-1662.
5. Golbe LI. The epidemiology of PSP. J Neural Transm 1994; 42(suppl):263-273.
6. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181-184.