20 year old Hispanic woman with bilateral blurry vision, nausea, weakness and severe headaches
Digital Journal of Ophthalmology 2005
Volume 11, Number 11
July 10, 2005
Volume 11, Number 11
July 10, 2005
Past Medical History:
- migraine headaches since age 12
Past Ocular History:
- visual auras with migraine
Social History:
- smokes ½ PPD
- no alcohol consumption
Family History:
- mother with migraine headaches
VA Count fingers OU
Pupils bilaterally reactive/no APD
EOM full OU
IOP 14 mmHg OD 12 mmHg OS
Anterior Segment
OD
2+ injection
1½+ cells
OS
1+ injection
2+ cells
Dilated Exam 1 + WBCs in vitreous OU
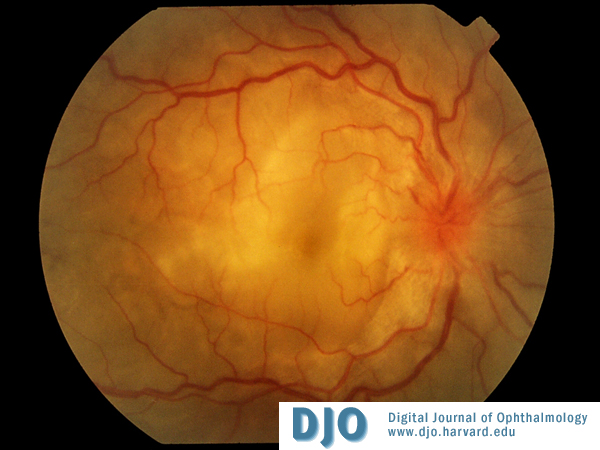
Color Photos OD
Bilateral bullous serous subretinal detachments and bilateral disc edema.
Bilateral bullous serous subretinal detachments and bilateral disc edema.
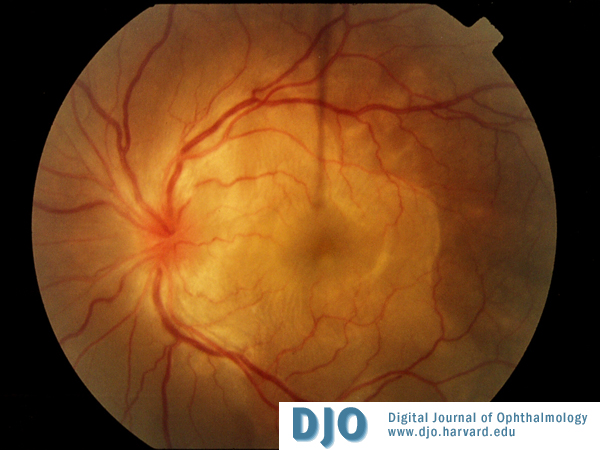
Color Photo OS
Bilateral bullous serous subretinal detachments and bilateral disc edema.
Bilateral bullous serous subretinal detachments and bilateral disc edema.
Serum
- Sedimentation Rate – 33 (normal range 0-15)
- ACE – normal
- Lysozyme – normal
- FTA-ABS – non-reactive
- RPR – non-reactive
- HLA-B27 negative
- HLA-DR4 positive
PPD – negative
Chest x-ray – unremarkable
Head CT/Head MRI – unremarkable
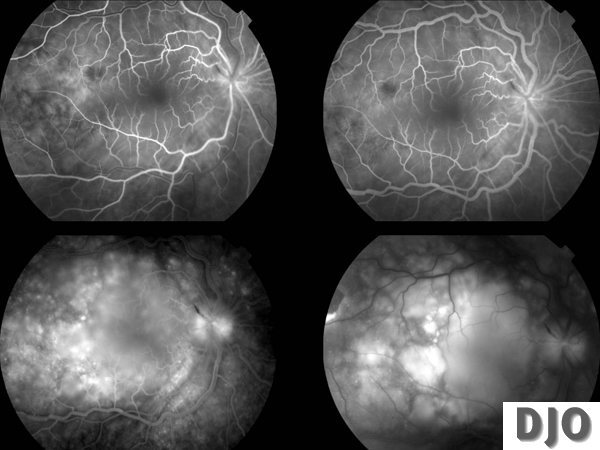
IVFA OD
Multiple pinpoint hyperfluorescent dots at the level of the retinal pigment epithelium in the early arteriovenous flow followed by pooling of fluorescein in the subretinal space, in the areas of exudative retinal detachment and optic nerve head leakage OU.
Multiple pinpoint hyperfluorescent dots at the level of the retinal pigment epithelium in the early arteriovenous flow followed by pooling of fluorescein in the subretinal space, in the areas of exudative retinal detachment and optic nerve head leakage OU.
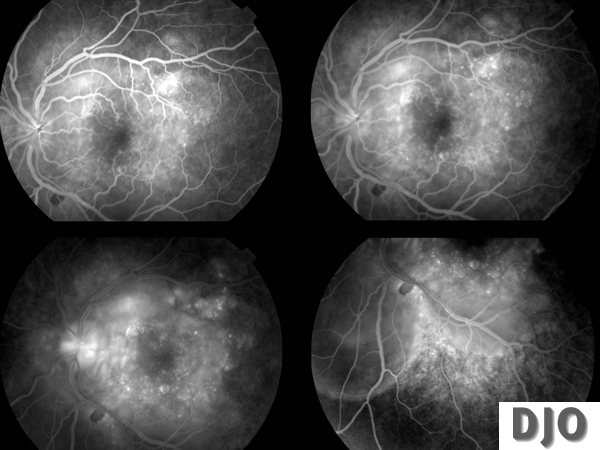
IVFA OS
Multiple pinpoint hyperfluorescent dots at the level of the retinal pigment epithelium in the early arteriovenous flow followed by pooling of fluorescein in the subretinal space, in the areas of exudative retinal detachment and optic nerve head leakage OU.
Multiple pinpoint hyperfluorescent dots at the level of the retinal pigment epithelium in the early arteriovenous flow followed by pooling of fluorescein in the subretinal space, in the areas of exudative retinal detachment and optic nerve head leakage OU.
• Uveal Effusion Syndrome
• Posterior Scleritis
• Primary Intraocular Lymphoma
• Sarcoidosis
• Syphilis
• Tuberculosis
Vogt-Koyanagi-Harada disease (VKH) is a systemic autoimmune disorder directed against antigens most likely associated with melanocytes present in such tissues as the choroid, the meninges, the inner ear, and the skin. It is characterized by bilateral, chronic, diffuse granulomatous uveitis, accompanied by characteristic neurological, auditory and integumentary features. The eye is typically the most involved organ, and visual sequelae are the most frequent and debilitating consequences of the disease.
Patients are typically 20 to 50 years old and have no history of either surgical or accidental ocular trauma. VKH affects mainly darkly pigmented races, such as East and Southeastern Asians, Asian Indians, Middle Easterners, Hispanics, and Native Americans; it is uncommon in Caucasians. VKH is also rare in Africans, suggesting that skin pigmentation alone is not the sole etiologic factor in its pathogenesis.
Based on revised diagnostic criteria, the disease is classified as complete, incomplete or probable based on the presence of extraocular findings (neurological, auditory and integumentary). No matter the form of the disease the patient manifests, three conditions must be present for the diagnosis of VKH: (1) no previous history of penetrating ocular trauma (either surgical or accidental); (2) no clinical or laboratory evidence suggestive of other ocular disease; and (3) bilateral ocular involvement.
The clinical course of VKH is divided in four phases: prodromal (mimics a viral infection), uveitic (bilateral diffuse uveitis with papillitis and exudative retinal detachment), convalescent (tissue depigmentation), and chronic recurrent (recurrent uveitis and ocular complications).
The pathogenesis of VKH is thought to be related to an aberrant T cell-mediated immune response directed against self-antigens found on melanocytes. VKH has been linked to human leukocyte antigen DR4 (HLA-DR4) and HLA-Dw53, with strongest associated risk for HLA-DRB1*0405 haplotype.
Comprehensive diagnostic criteria for VKH were revised during the 2001 First International Workshop on Vogt-Koyanagi-Harada Disease. The diagnosis of VKH is clinical, and differential includes sympathetic ophthalmia, sarcoidosis, primary intraocular B-cell lymphoma, posterior scleritis, and uveal effusion syndrome.
The standard initial therapy for VKH is prompt and aggressive use of systemic corticosteroids, to which the disease is especially responsive, particularly in the early stages. Typical dosing regimens range from 1.0 to 2.0 mg/kg/day of oral prednisone or 200 mg of intravenous methylprednisolone for 3 days, followed by or high-dose corticosteroids. Early therapy with a slow taper based on clinical response, usually over a minimum of 6 months, has been shown to improve the prognosis by reducing the length of disease, increasing the incidence of a convalescent phase, and decreasing the extraocular manifestations. Other immunomondulaory agents (most oftentimes cyclosporine) may be needed for non-responsive patients or when corticosteroid side-effects are not tolerated. Visual prognosis with VKH is generally good with prompt diagnosis and aggressive immunomodulatory treatment.
Snyder DA, Tessler HH. Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol 1980;90:69-75.
Nussenblatt RB. Clinical studies of Vogt-Koyanagi-Harada's disease at the National Eye Institute, NIH, USA. Jpn J Ophthalmol 1988;32:330-3.
Rubsamen PE, Gass JD. Vogt-Koyanagi-Harada syndrome. Clinical course, therapy, and long-term visual outcome. Arch Ophthalmol 1991;109:682-7.
Ohno S, Minakawa R, Matsuda H. Clinical studies of Vogt-Koyanagi-Harada's disease. Jpn J Ophthalmol 1988;32:334-43.
Sasamoto Y, Ohno S, Matsuda H. Studies on corticosteroid therapy in Vogt-Koyanagi-Harada disease. Ophthalmologica 1990;201:162-7.
Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Pivetti-Pezzi P, Tessler HH, Usui M. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 2001;131:647-52.
Kamondi A, Szegedi A, Papp A, Seres A, Szirmai I. Vogt-Koyanagi-Harada disease presenting initially as aseptic meningoencephalitis. Eur J Neurol 2000;7:719-22.
Goto H, Rao NA. Sympathetic ophthalmia and Vogt-Koyanagi-Harada syndrome. Int Ophthalmol Clin 1990;30:279-85.
Lubin JR, Ni C, Albert DM. Clinicopathological study of the Vogt-Koyanagi-Harada syndrome. Int Ophthalmol Clin 1982l;22:141-56.
Shindo Y, Inoko H, Yamamoto T, Ohno S. HLA-DRB1 typing of Vogt-Koyanagi-Harada's disease by PCR-RFLP and the strong association with DRB1*0405 and DRB1*0410. Br J Ophthalmol 1994;78:223-6.
Damico FM, Cunha-Neto E, Goldberg AC, Iwai LK Marin ML, Hammer J, Kalil J, Yamamoto JH. T cell recognition and cytokine profile induced by melanocyte epitopes in HLA-DRB1*0405-positive and -negative Vogt-Koyanagi-Harada uveitis patients. Invest Ophthalmol Vis Sci. [in press]2005.
Chan CC, Palestine AG, Nussenblatt RB, Roberge FG, Benezra D. Anti-retinal auto-antibodies in Vogt-Koyanagi-Harada syndrome, Behcet's disease, and sympathetic ophthalmia. Ophthalmology 1985;92:1025-8.
Moorthy RS, Inomata H, Rao NA. Vogt-Koyanagi-Harada syndrome. Surv Ophthalmol 1995;39:265-92.
Oshima Y, Harino S, Hara Y, Tano Y. Indocyanine green angiographic findings in Vogt-Koyanagi-Harada disease. Am J Ophthalmol 1996;122:58-66.
Read RW, VKH Therapy Group. Poster 2664: A retrospective multinational survey of current treatment patterns in acute and subacute Vogt-Koyanagi-Harada disease. 2004 Association for Research in Vision and Ophthalmology Annual Meeting. Fort Lauderdale, FL, April 25-29.
Paredes IP, Ahmed M, Foster CS. Poster 2663: Immunomodulatory therapy for VKH patients as a first line therapy. 2004 Association for Research in Vision and Ophthalmology Annual Meeting. Fort Lauderdale, FL, April 25-29.
Wakatsuki Y, Kogure M, Takahashi Y, Oguro Y. Combination therapy with cyclosporin A and steroid in severe case of Vogt-Koyanagi-Harada's disease. Jpn J Ophthalmol 1988;32:358-60.
Read RW, Rechodouni A, Butani N, Johnston R, LaBree LD, Smith RE, Rao NA. Complications and prognostic factors in Vogt-Koyanagi-Harada disease. Am J Ophthalmol 2001;131:599-606.