A 20-year-old woman with abnormal eye movements
Digital Journal of Ophthalmology 2021
Volume 27, Number 1
January 4, 2021
Volume 27, Number 1
January 4, 2021
Download PDF
Vivian Paraskevi Douglas, MD, DVM, MBA | Department of Ophthalmology, Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, Massachusetts
Homer H. Chiang, MD | Department of Ophthalmology, UT Health San Antonio, San Antonio, Texas
Konstantinos A. A. Douglas, MD, DVM, MBA | Department of Ophthalmology, Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, Massachusetts
Tavé Van Zyl, MD | Department of Ophthalmology, Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, Massachusetts
Nurhan Torun, MD | Division of Ophthalmology, Department of Surgery, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts
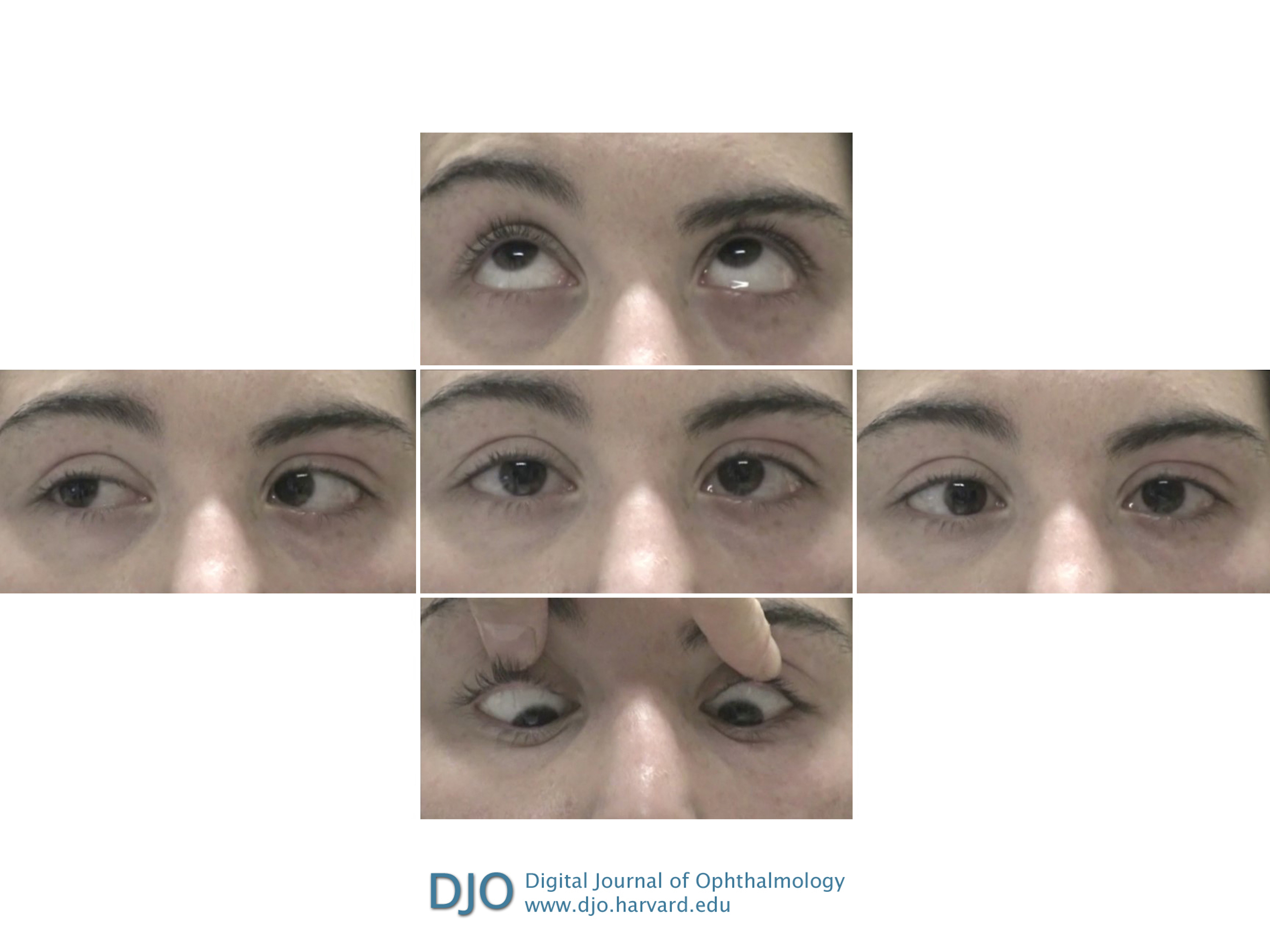
Figure 1
Clinical photographs of extraocular motility examination in a 20-year-old woman who presented with oblique binocular diplopia showing impaired leftward gaze in both eyes (middle row, right). Although right gaze appears intact (middle row, left), she had disconjugate eye movements, with slow adducting saccades in the left eye.
Clinical photographs of extraocular motility examination in a 20-year-old woman who presented with oblique binocular diplopia showing impaired leftward gaze in both eyes (middle row, right). Although right gaze appears intact (middle row, left), she had disconjugate eye movements, with slow adducting saccades in the left eye.
Video 1
Video of patient showing convergence at 1-2 seconds, right gaze at 5 seconds, attempted left gaze with leftward gaze palsy at 10-11 seconds, upgaze at 14 seconds, downgaze at 17 seconds, and a slowly adducting saccade in the left eye, indicating left internuclear ophthalmoplegia, at 21 seconds. Additionally, at 12 and 19 seconds the patient blinks, clearly showing lagophthalmos in the left eye due to left facial nerve palsy.
Video of patient showing convergence at 1-2 seconds, right gaze at 5 seconds, attempted left gaze with leftward gaze palsy at 10-11 seconds, upgaze at 14 seconds, downgaze at 17 seconds, and a slowly adducting saccade in the left eye, indicating left internuclear ophthalmoplegia, at 21 seconds. Additionally, at 12 and 19 seconds the patient blinks, clearly showing lagophthalmos in the left eye due to left facial nerve palsy.
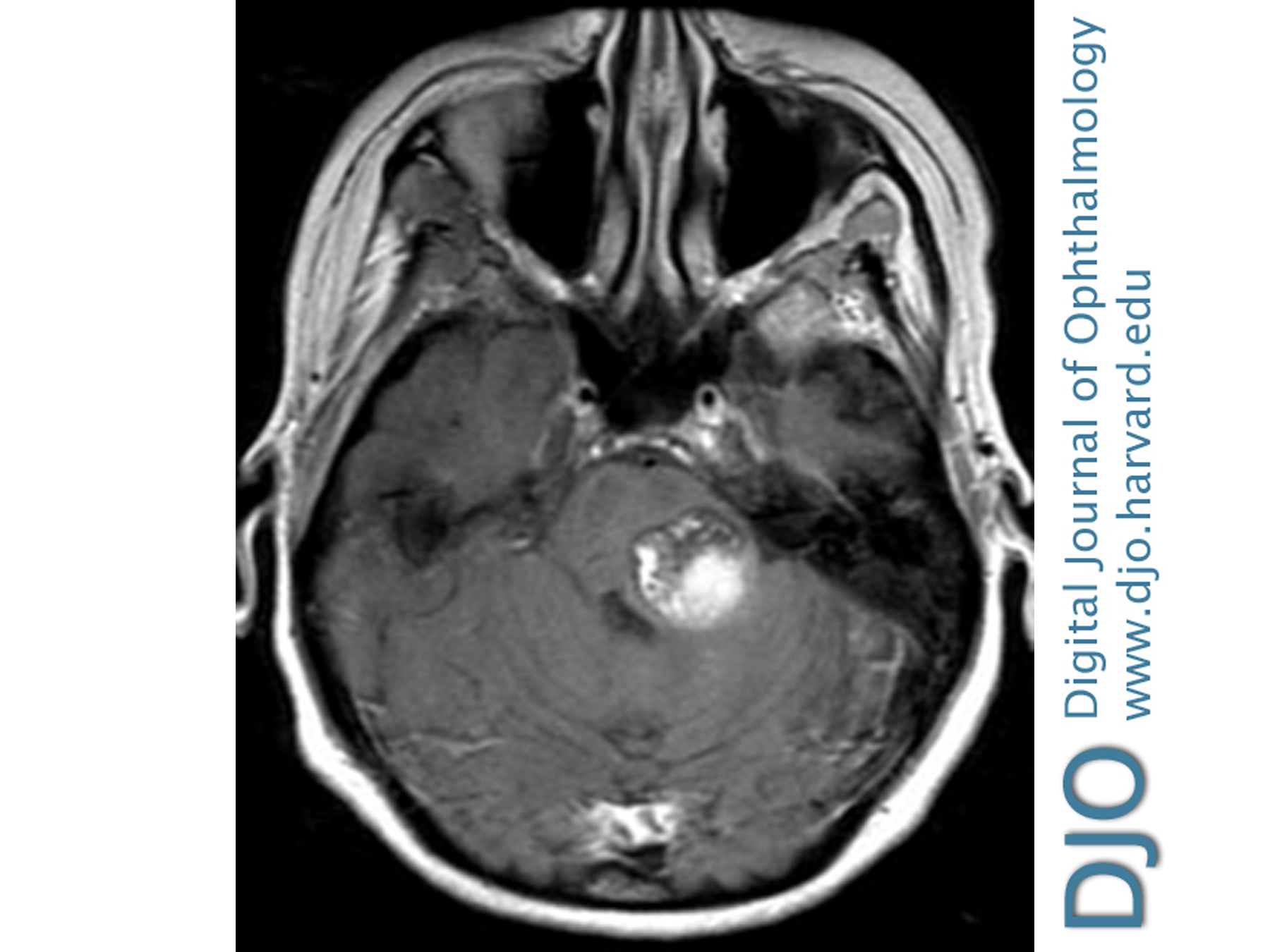
Figure 2
T1-weighted axial magnetic resonance imaging demonstrating a “popcorn” hyperintensity in the left posterior pons, with adjacent hypointensities anterior and anteromedial to the lesion consistent with remote hemorrhage, all in keeping with left pontine cavernous malformation.
T1-weighted axial magnetic resonance imaging demonstrating a “popcorn” hyperintensity in the left posterior pons, with adjacent hypointensities anterior and anteromedial to the lesion consistent with remote hemorrhage, all in keeping with left pontine cavernous malformation.
In addition, lubricating drops, four times a day, lubricating ointment at bedtime, and humidifiers were recommended because of underlying exposure keratopathy. Two weeks after surgery, however, she had an esotropia of 6 prism diopters in primary position at distance, without tropia at near. Fresnel stick-on prism lenses were prescribed. Overall, no improvement on either the motility or facial nerve function was noted postoperatively.
The syndrome can be expanded based on involvement of additional brainstem structures. Additional facial nerve deficit may be present as fibers course around the abducens nerve nucleus before exiting the brainstem. When this occurs, the condition is termed eight-and-a-half syndrome, as was present in our patient.(2) Another example is the addition of ipsilateral trigeminal nerve involvement that produces a thirteen-and-a-half syndrome variant.(3)
These syndromes, as in our patient, result from a unilateral lesion in the dorsal pontine tegmentum. The differential diagnosis includes ischemia (eg, pontine lacunar infarction), compressive lesion (eg tumor), demyelinating disease, and infection.3 Thorough medical history and physical examination along with neuroimaging are key to early diagnosis and treatment of these syndromes, because the prognosis is variable and the severity of the disorder is determined largely by the specifics of the underlying pathology.
On imaging, these lesions can resemble gliomas, hemorrhagic telangiectasias, such as those seen in Osler-Weber-Rendu disease, and even brain metastasis.(4)
Cavernous malformations (CMs) are well-defined, tightly packed masses of thin-walled, dilated, sinusoidal capillaries with endothelial lining and fibrous adventitia. Although CMs may occur anywhere in the central nervous system, most are found in the cerebrum, accounting for up to 25% of all vascular malformations. In a meta-analysis of 11 natural history studies, brainstem CMs comprised 18 % of lesions.(6)
Because of the high density of cranial nerve nuclei and associated nerve tracts, patients with brainstem CMs commonly present with multiple cranial neuropathies and variable ophthalmic manifestations. Progressive neurological deficits are not uncommon and were observed in 39 % of patients in one case series.(1)
Characteristic T1- and T2-weighted MRI findings include a “popcorn” pattern of variable intensity due to evolving blood products. Remote bleeds may appear as a hemosiderin ring in the periphery of the lesion.
In conclusion, brainstem CMs can have variable ophthalmic manifestations owing to the close proximity of cranial nerve nuclei in the brainstem. Common ophthalmic manifestations include extraocular motility deficits that result in diplopia and long-term ocular surface sequelae due to lagophthalmos, with decreased or absent blink. Early recognition is of key importance for determining proper treatment of brainstem CMs, which should include the input of neurologists, neuro-ophthalmologists, and neurosurgeons for optimal results.
2. Eggenberger E. Eight-and-a-half syndrome: one-and-a-half syndrome plus cranial nerve VII palsy. J Neuroophthalmol 1998;18:114-6.
3. Allbon DS, La Hood B. Thirteen-and-a-half syndrome. J Neuroophthalmol 2016;36:191-2.
4. Mouchtouris N, Chalouhi N, Chitale A, et al. Management of cerebral cavernous malformations: from diagnosis to treatment ScientificWorldJournal 2015;2015:808314.
5. Mesina BVQ, Sosuan GMN, Reyes KB. Eight-and-a-half syndrome: a rare potentially life-threatening disease. GMS Ophthalmol Cases 2018;8:Doc04.
6. Gross BA, Lin N, Du R, Day AL. The natural history of intracranial cavernous malformations. Neurosurg Focus 2011;30:E24.