A 10-year-old girl with multiple eyelid neuroproliferative tumors
Digital Journal of Ophthalmology 2021
Volume 27, Number 3
July 12, 2021
Volume 27, Number 3
July 12, 2021
Download PDF
Kristin Torroella | George Washington School of Medicine and Health Sciences, Washington, DCGeorge Washington School of Medicine and Health Sciences, Washington, DC
Jana Bregman, MD | Department of Ophthalmology, Childrens National Hospital, Washington, DC
Maria Isabel Almira-Suarez, MD, FASCP | Neuropathology Service, Division of Anatomic Pathology, Childrens National Hospital, Washington, DC
Marijean Miller, MD | Department of Ophthalmology, Childrens National Hospital, Washington, DC
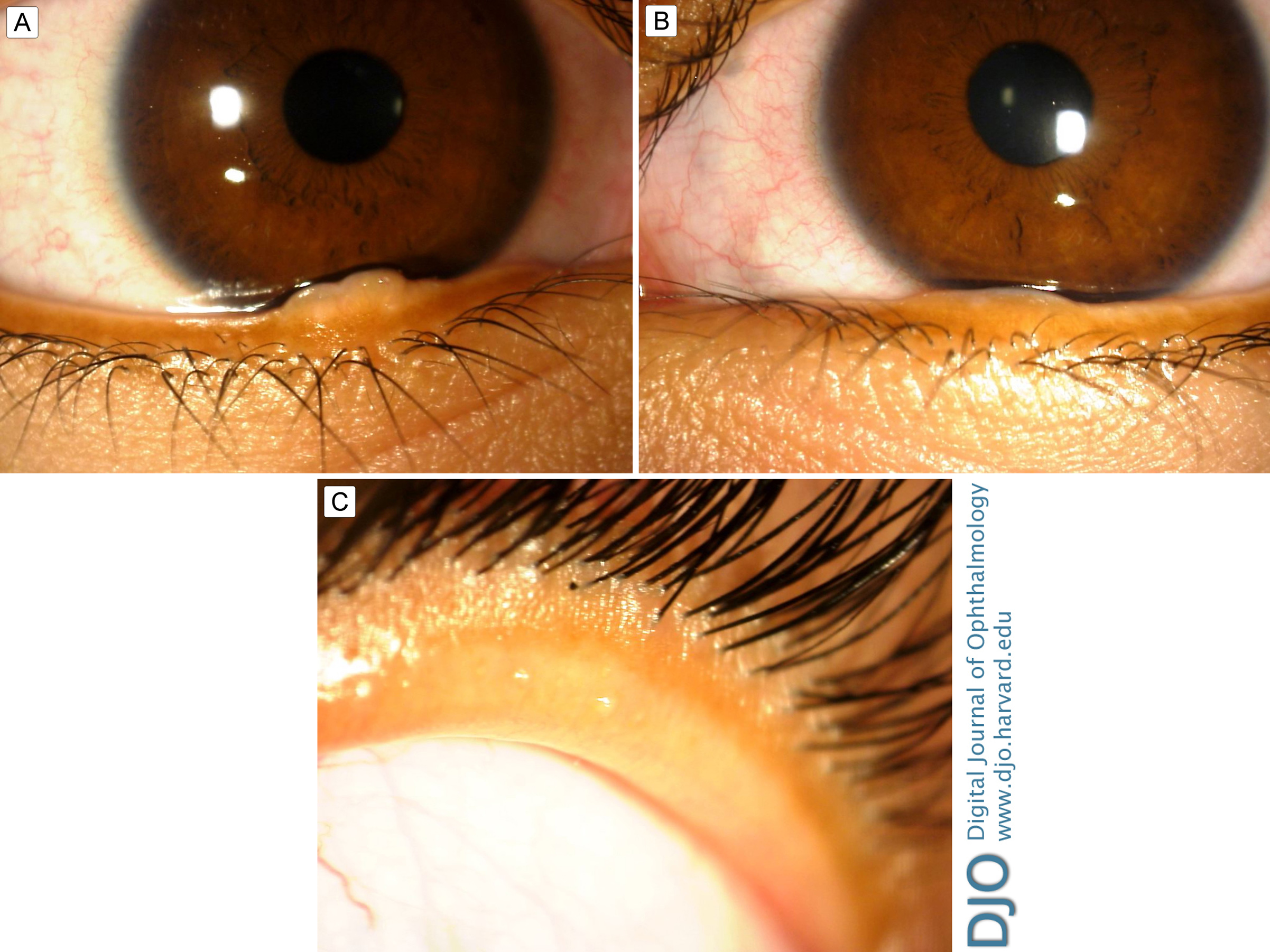
Figure 1.
A, Right lower lid margin showing a 4.1 mm gelatinous, flesh-colored, ovoid nodule. B, Left lower lid margin showing a 4.4 mm gelatinous, flesh-colored nodule. C, Thickened right upper eyelid to 5 mm.
A, Right lower lid margin showing a 4.1 mm gelatinous, flesh-colored, ovoid nodule. B, Left lower lid margin showing a 4.4 mm gelatinous, flesh-colored nodule. C, Thickened right upper eyelid to 5 mm.
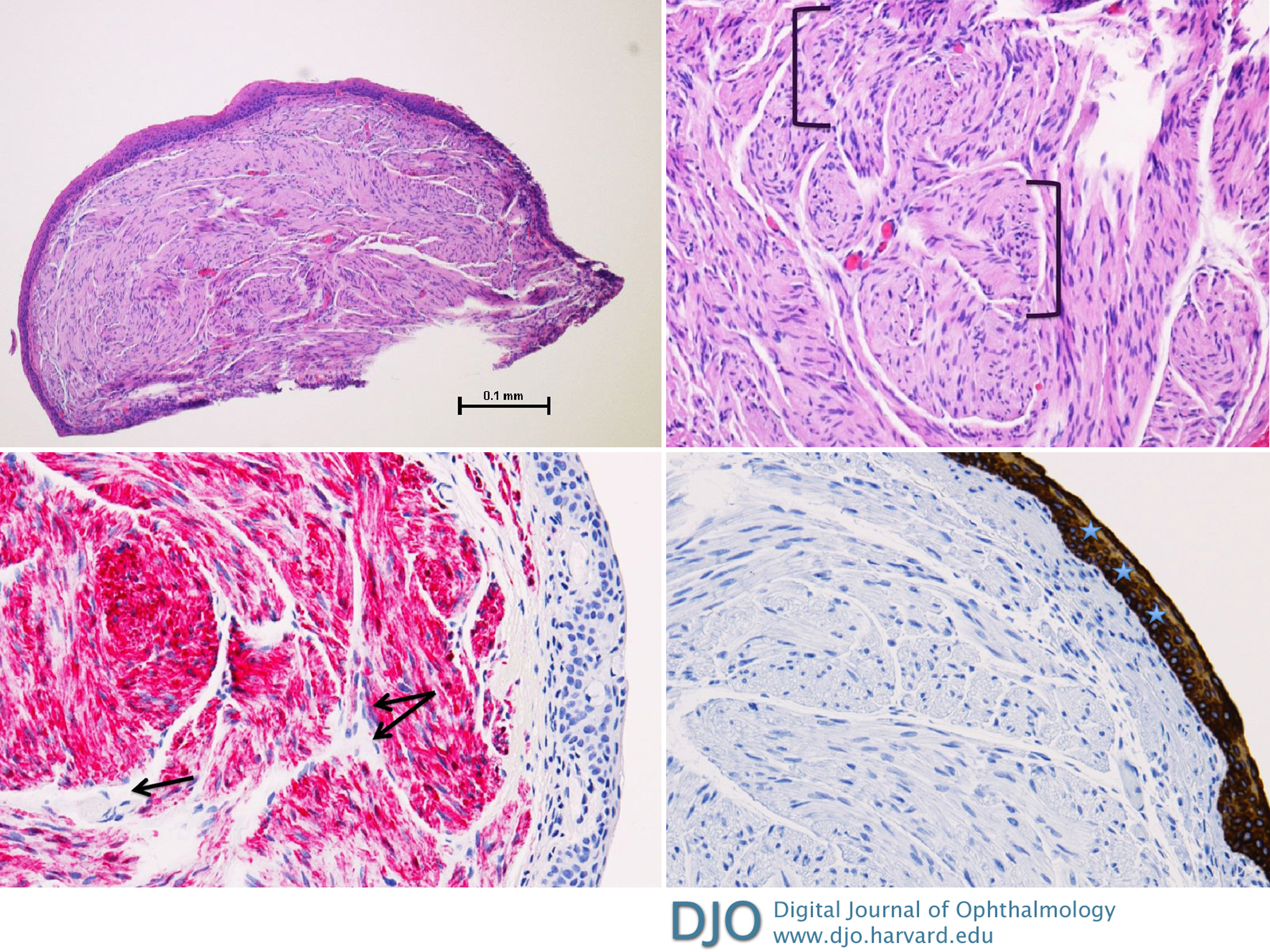
Figure 2
A, Plexiform schwannoma (hematoxylin-eosin [H&E], original magnification ×4). B, Eye lesion highlighting disorganized nerve pattern (H&E, original magnification ×10). C, Positive S100 immunohistochemical stain highlighting the neural origin of the lesion (original magnification ×20). D, Pan-cytokeratin stain highlighting epithelial tissue (original magnification ×20).
A, Plexiform schwannoma (hematoxylin-eosin [H&E], original magnification ×4). B, Eye lesion highlighting disorganized nerve pattern (H&E, original magnification ×10). C, Positive S100 immunohistochemical stain highlighting the neural origin of the lesion (original magnification ×20). D, Pan-cytokeratin stain highlighting epithelial tissue (original magnification ×20).
Malignant eyelid lesions are generally rare in the pediatric population. Examples include basal cell carcinoma, squamous cell or sebaceous cell carcinoma, and keratoacanthoma, which is categorized as a premalignant lesion. Malignant features such as sentinel vessels, bleeding or ulceration, distortion of the lid margin, or madarosis were all absent in this case.
With genetic testing becoming more available, it is now common to screen newborns with a family history of MEN2B, allowing for earlier molecular diagnosis and thus improved outcomes.(10) However, 50% of MEN2B cases stem from de novo mutations, resulting in delayed diagnosis and poorer prognosis.(10) In nonfamilial MEN2B, patients are most often diagnosed at an advanced stage, triggered by systemic symptoms related to elevated catecholamines due to pheochromocytoma or elevated calcitonin due to MTC.(6) By this point in time, MTC has often progressed to an advanced stage, marked by metastatic cancer most commonly seen in the liver and lungs.(8)
Early MEN2B diagnosis allows for prompt MTC surveillance, surgical resection of the tumor or even prophylactic thyroidectomy if needed, which can be life-saving.(11) Early diagnosis of asymptomatic patients with de novo mutations is challenging and relies on clinical findings, including ocular abnormalities.(12) Ocular changes may be the first manifestations of MEN2B, presenting before systemic signs and symptoms of pheochromocytoma and/or MTC.(6,11) In addition, ocular findings can be detected with a noninvasive examination, whereas identifying other systemic findings in MEN2B may require imaging, lab work, and even diagnostic biopsies. Although the ophthalmic signs are not visually significant, they can play a critical role in early detection.(11) The exact incidence of ocular findings is unknown since MEN2B itself is rare.(11) However, of 33 reported ocular cases, 100% of patients were found to have prominent corneal nerves. Additional findings included eyelid neural tumors or thickening (88%), subconjunctival neuromas (79%), decreased tear production (48%), rostral displacement of the cilia (12%), and poor pupillary dilation (12%).(11)
Our patient was found to have prominent corneal nerves, thickened eyelids, and multiple eyelid conjunctival plexiform schwannomas. Histologic studies have demonstrated that the appearance of prominent corneal nerves is due to nonmyelinated axons associated with Schwann cells.(13) Although prominent corneal nerves have been reported in 100% of MEN2B cases, they can also be seen in a number of other diseases such as MEN2A, Refsum disease, pheochromocytoma, primary amyloidosis, ectodermal dysplasia, leprosy and herpes zoster keratopathy.(6,9) Therefore, it is important to be aware of additional distinctive ocular features of MEN2B, such as eyelid thickening, ptosis, impaired pupillary dilation, and eyelid, conjunctival, and subconjunctival neuroproliferative tumors.(11)
Plexiform schwannomas—also referred to as neurilemomas, neurolemomas, or neuromas—are benign tumor proliferations of nerve tissue. In MEN2B, they are seen as multiple unencapsulated lesions in the palpebral conjunctiva or subconjunctiva.(14) A diagnostic biopsy can help confirm the suspected diagnosis of MEN2B associated neural lesion, which reacts positively with the immunohistochemical S100 and demonstrates a disorganized conglomeration of nerve twigs.(10,11) The formation of nodules is attributed to prominent nerves forming plexiform bundles aligned parallel to the epidermis.(6,7,14) These lesions often increase in size over time, which in many patients, as in ours, prompt further ophthalmologic evaluation.(10)
Such tumors can be surgically removed under local anesthesia with a shave biopsy or full-thickness wedge resection under monitored sedation.(15) However, with the former, there is a risk of residual tumor being left behind, possibly leading to reocurrence.(14,15) In order to prevent eyelid notching during direct closure of the lid defect following wedge resection, the tarsus and eyelid margin are meticulously reapproximated.(15) If the lesion is large (greater than 30% of the eyelid width), a more complex eyelid reconstruction is necessary, requiring tarsoconjunctival and skin grafts from adjacent tissues to reconstruct the anterior and posterior lamellae of the eyelid.
This pediatric case of MEN2B highlights important ocular findings in MEN2B, some of which can be detected by the naked eye, with no need for a slit-lamp examination or invasive testing. Ophthalmologists aware of its ocular manifestations can play a vital role in detecting MEN2B at an early precancerous stage, establishing care with other subspecialists as needed (ie, endocrinology), and helping to screen immediate family members who may also be affected.
2. Castillo BV Jr, Kaufman L. Pediatric tumors of the eye and orbit. Pediatr Clin North Am 2003;50:149-72.
3. 3. Ozer PA, Gurkan A, Kurtul BE, Kabatas EU, Beken S. Comparative clinical outcomes of pediatric patients presenting with eyelid nodules of idiopathic facial aseptic granuloma, hordeola, and chalazia. J Pediatr Ophthalmol Strabismus 53.4 2016:206-11.
4. Sharma S, Panda A, Jana M, Arora A, Sharma SK. Ophthalmic manifestations of systemic diseases—part 1: phakomatoses, hematologic malignancies, metastases, and histiocytosis. Curr Probl Diagn Radiol 2014;43:175-85.
5. Cannon T, Carter K, Folberg R. Neurogenic tumors of the orbit. Duane’s Clinical Ophthalmology. CD-ROM edition. Vol. 2, chap. 41. Philadelphia: Lippincott Williams & Wilkins, 2006.
6. Sahin A, Yildirim N. Ocular findings in a child with multiple endocrine neoplasia type 2b. J Pediatr Ophthalmol Strabismus 2008;45:313-5.
7. Eter N, Klingmüller D, Höppner W, Spitznas M. Typical ocular findings in a patient with multiple endocrine neoplasia type 2b syndrome. Graefes Arch Clin Exp Ophthalmol 2001;239:391-4.
8. Majidi M, Haghpanah V, Hedayati M, Khashayar P, Mohajeri-Tehrani MR, Larijani B. A family presenting with multiple endocrine neoplasia type 2B: a case report. J Med Case Rep 2011;5:587.
9. Parker DG, Robinson BG, O’Donnell BA. External ophthalmic findings in multiple endocrine neoplasia type 2B. Clin Exp Ophthalmol 2004;32:420-3.
10. Puvanachandra N, Aroichane M. Diffuse corneoscleral limbal neuromas with prominent corneal nerves in multiple endocrine neoplasia syndrome type IIB. J Pediatr Ophthalmol Strabismus 2010;47:171-3.
11. Jacobs JM, Hawes MJ. From eyelid bumps to thyroid lumps: report of a MEN type IIb family and review of the literature. Ophthalmic Plast Reconstr Surg 2001;17:195-201.
12. Leboulleux S, Travagli JP, Caillou B, et al. Medullary thyroid carcinoma as part of a multiple endocrine neoplasia type 2B syndrome: influence of the stage on the clinical course. Cancer 2002;94:44-50.
13. Fink A, Lapidot M, Spierer A. Ocular manifestations in multiple endocrine neoplasia type 2b. Am J Ophthalmol 1998;126:305-7.
14. Jakobiec F, Rashid A, Yoon M. Isolated nonsyndromic intraneural neuroma of the eyelid skin. Ophthal Plast Reconstr Surg 2016;32:e147-9.
15. Mehta S, Belliveau MJ, Oestreicher JH. Oculoplastic surgery. Clin Plast Surg 2013;40:631-51.