A 41-year-old man with bilateral, painless loss of vision
Digital Journal of Ophthalmology 2021
Volume 27, Number 4
November 5, 2021
Volume 27, Number 4
November 5, 2021
Download PDF
Michael Chang, BS | School of Medicine, University of Maryland, Baltimore, Maryland
Kin K. Yee, MD | Department of Ophthalmology, Georgetown University, Washington, DC
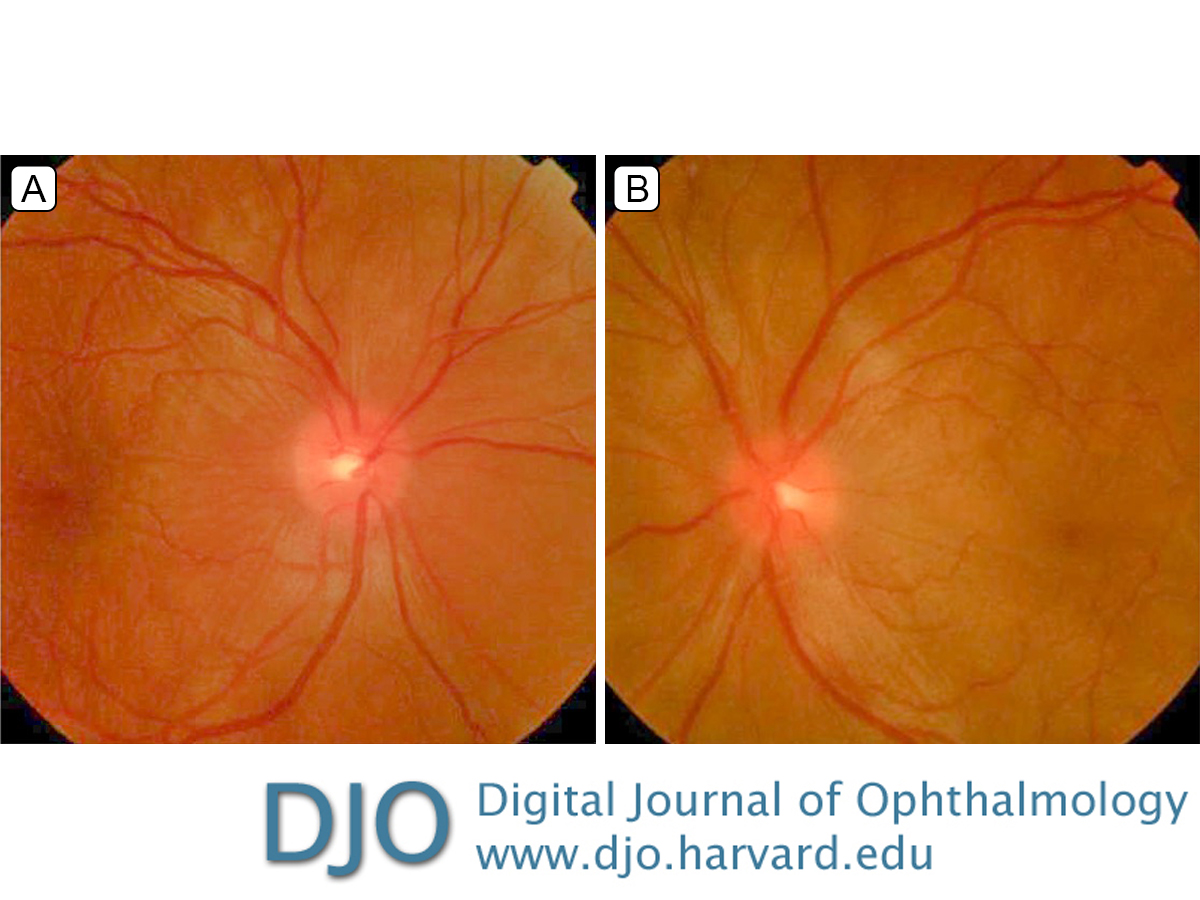
Figure 1.
Fundus photographs of the right eye (A), showing mild optic nerve edema, retinal striae, and localized subretinal fluid in the macula, and the left eye (B), showing mild optic nerve edema and serous retinal detachments in the macula and around the optic nerve.
Fundus photographs of the right eye (A), showing mild optic nerve edema, retinal striae, and localized subretinal fluid in the macula, and the left eye (B), showing mild optic nerve edema and serous retinal detachments in the macula and around the optic nerve.
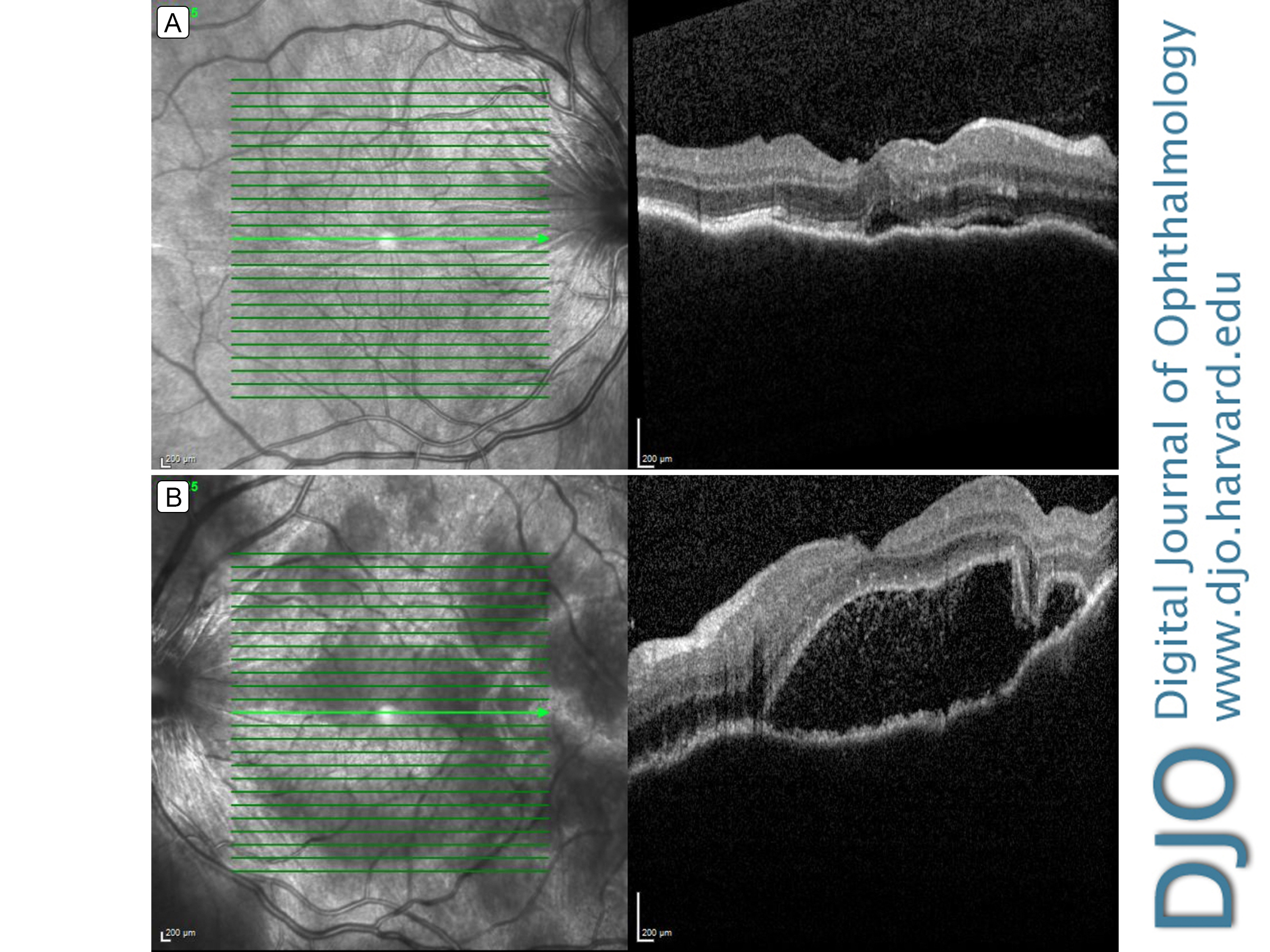
Figure 2.
Retinal optical coherence tomography (OCT) showing choroidal folds and subretinal fluid in the right eye (A) and choroidal folds, loculated pockets of subretinal fluid, and hyper-reflective dots in the subretinal space in the left eye (B).
Retinal optical coherence tomography (OCT) showing choroidal folds and subretinal fluid in the right eye (A) and choroidal folds, loculated pockets of subretinal fluid, and hyper-reflective dots in the subretinal space in the left eye (B).
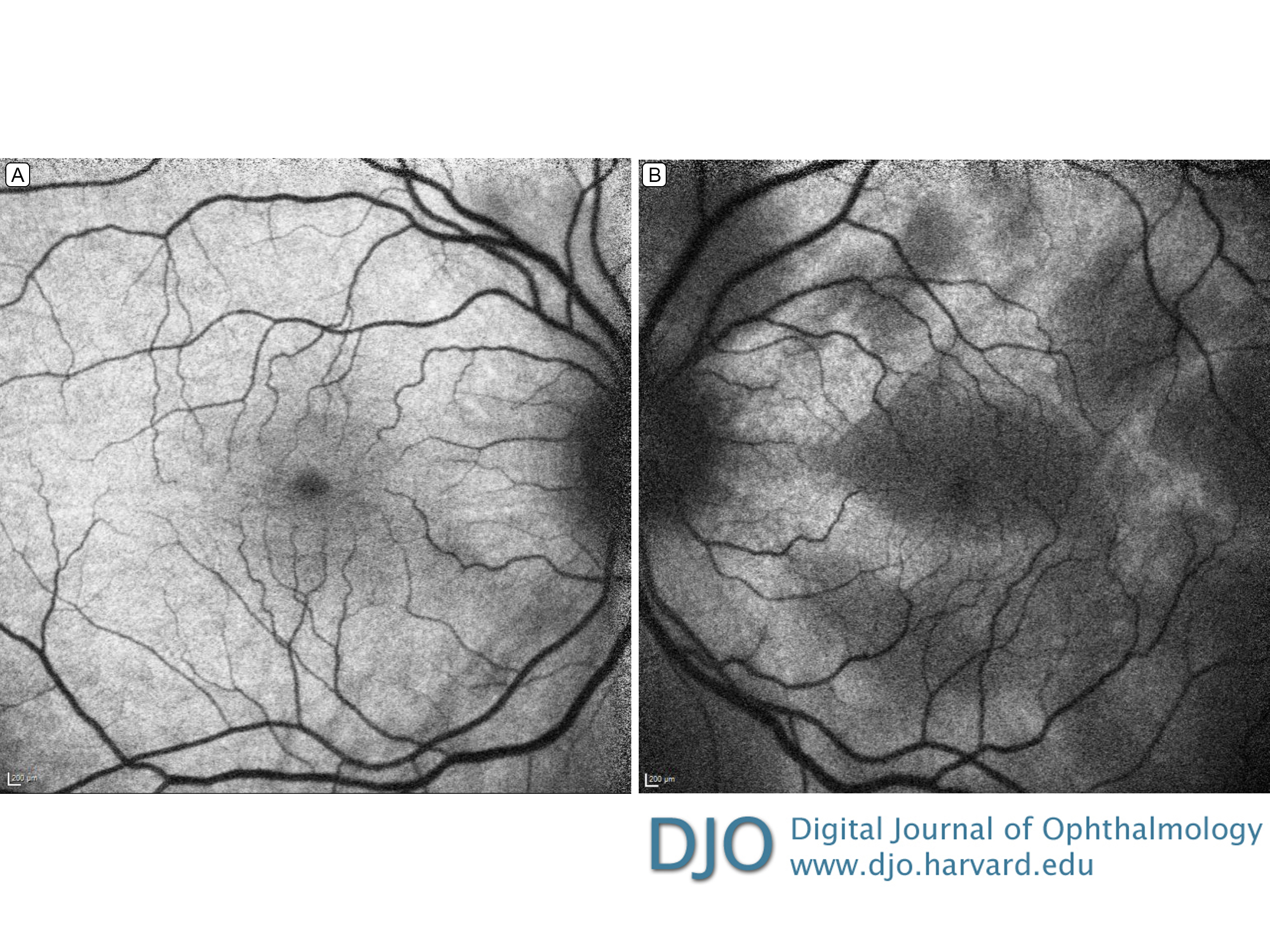
Figure 3.
Fundus autofluorescence. The right eye (A) shows mild hypo-autofluorescence around the fovea and optic nerve; the left eye (B), patches of hypo-autofluorescence corresponding to pockets of serous detachments in the macula.
Fundus autofluorescence. The right eye (A) shows mild hypo-autofluorescence around the fovea and optic nerve; the left eye (B), patches of hypo-autofluorescence corresponding to pockets of serous detachments in the macula.
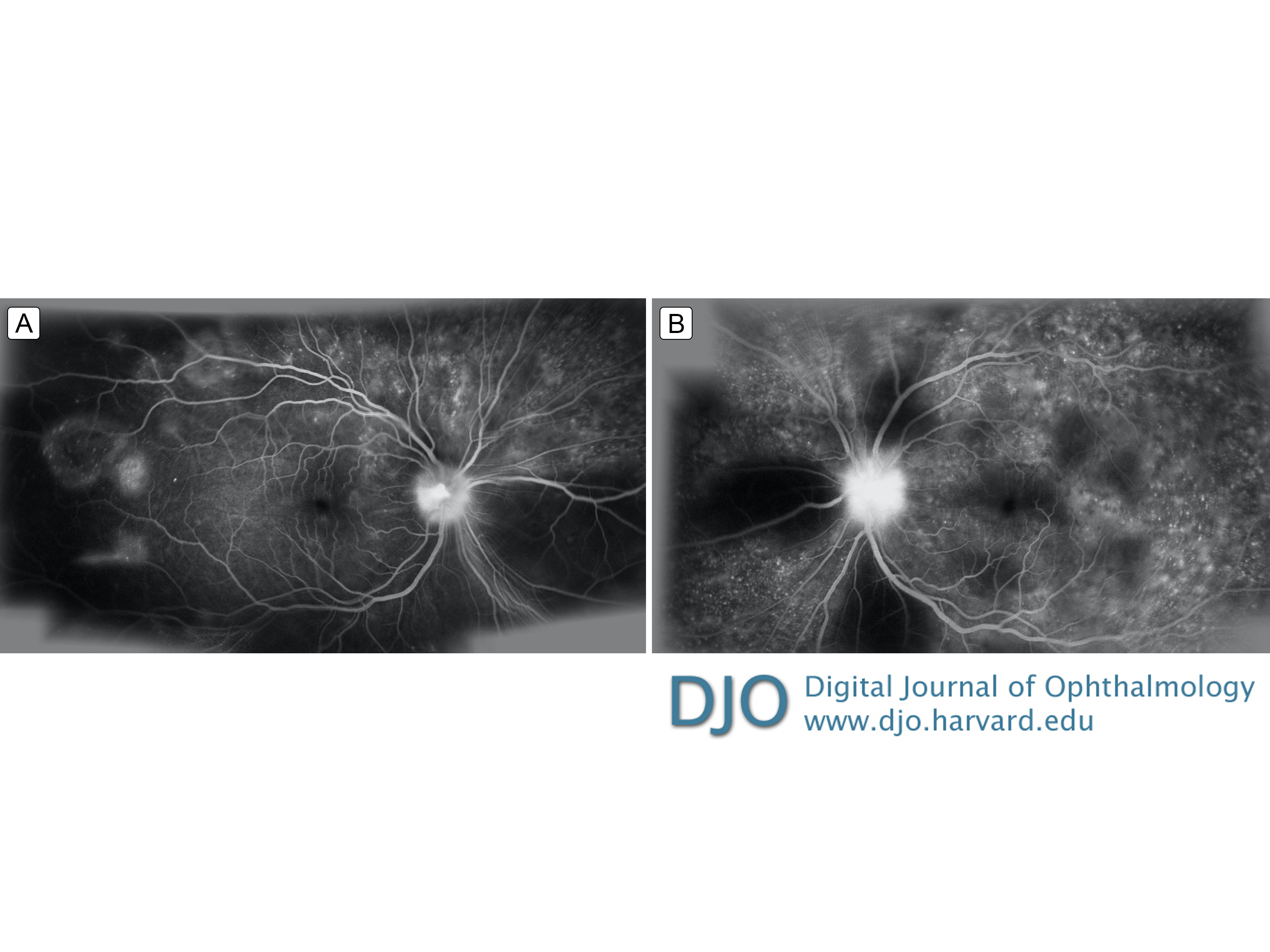
Figure 4.
Late-phase fluorescein angiography showing multiple pinpoint foci of leakage in the macula and retinal periphery with optic nerve leakage, greater in the left eye (B) than in the right eye (A). There are areas of hypo-fluorescence due to blockage from serous detachments.
Late-phase fluorescein angiography showing multiple pinpoint foci of leakage in the macula and retinal periphery with optic nerve leakage, greater in the left eye (B) than in the right eye (A). There are areas of hypo-fluorescence due to blockage from serous detachments.
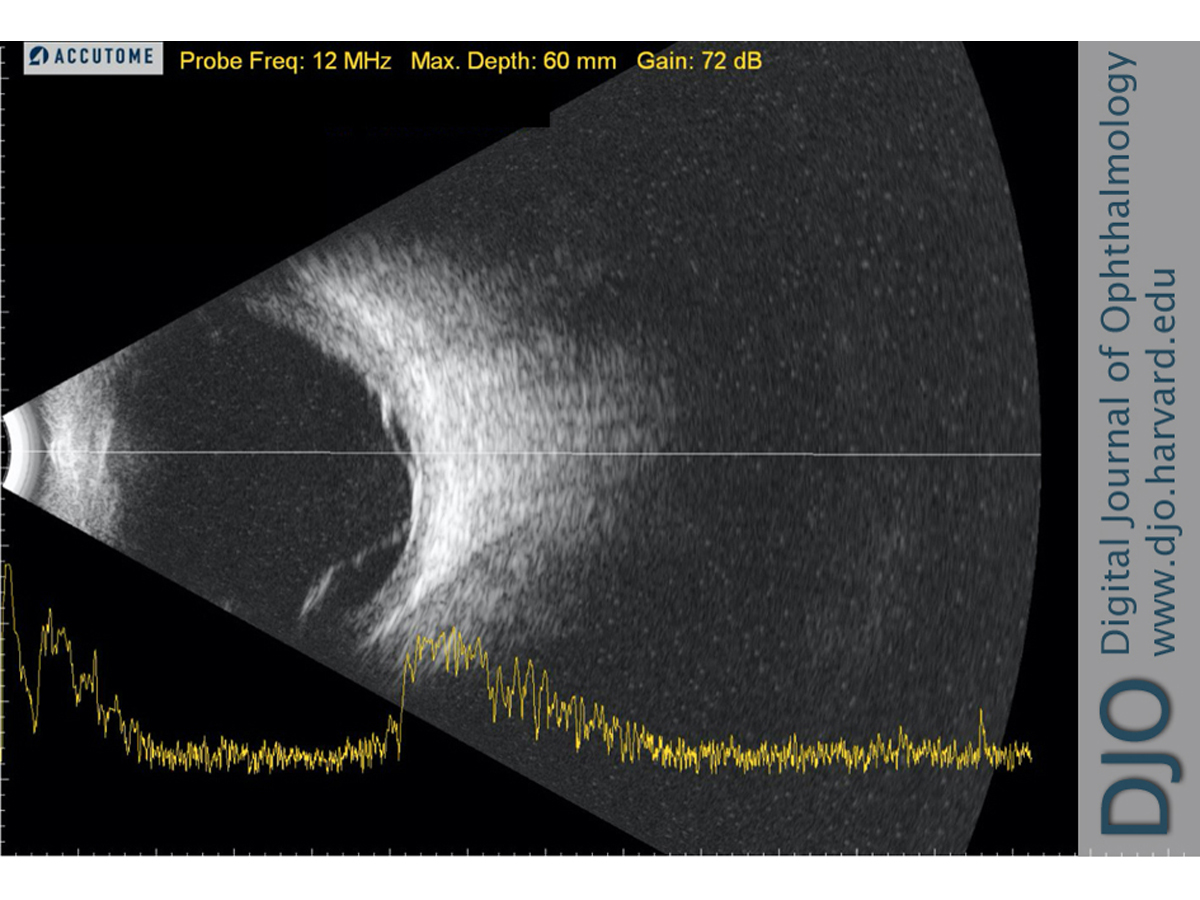
Figure 5.
B-scan ultrasound of the left eye showing a thickened choroid and exudative retinal detachments; there was no evidence of the classic T sign seen in posterior scleritis.
B-scan ultrasound of the left eye showing a thickened choroid and exudative retinal detachments; there was no evidence of the classic T sign seen in posterior scleritis.

Figure 6.
Retinal OCT 10 weeks after treatment demonstrates resolution of subretinal fluid in both the right (A) and left (B) eyes; the left eye shows patchy loss of the ellipsoid zone.
Retinal OCT 10 weeks after treatment demonstrates resolution of subretinal fluid in both the right (A) and left (B) eyes; the left eye shows patchy loss of the ellipsoid zone.
VKH more often affects people with darker skin pigmentation, including Asian, Hispanic, Native American, and Middle Eastern ethnic groups.(1,3) The incidence of VKH is variable, accounting for approximately 4% of uveitis cases in the United States and 8% in Japan.(3) Most studies report a slight female preponderance. (1) Affected individuals typically range in age from 20 to 50 years.(4)
To diagnose VKH, patients must not have a history of ocular trauma or eye surgery. The disease is classified as (1) complete, (2) incomplete, or (3) probable. Complete disease is associated with bilateral ocular, neurologic, auditory, and integumentary symptoms. Patients with incomplete VKH have bilateral ocular involvement and neurologic, auditory, or skin findings. Patients with probable VKH present with only bilateral ocular symptoms.(1)
VKH is usually diagnosed based on clinical appearance. Patients almost always present with bilateral exudative retinal detachments, although there may be asymmetry between the eyes. Fluorescein angiography shows multiple punctate hyperfluoresecent foci, with pooling in areas of serous retinal detachment.(3) Focal areas of retinal pigment epithelium loss may give rise to window defects, and optic disc leakage is often present. Indocyanine green angiography demonstrates delayed choroidal perfusion, with multiple hypercyanescent foci of leakage in areas of exudative retinal detachment.(3) Retinal OCT findings include exudative detachments, which often form loculated pockets of subretinal fluid, choroidal excavations, the presence of fibrin in the subretinal space and a thickened choroid. B-scan ultrasound demonstrates choroidal thickening with or without an exudative retinal detachment. Cerebrospinal fluid may show lymphocytic pleocytosis.
There are four stages of VKH: (1) prodromal, (2) acute uveitic, (3), convalescent, and (4) chronic recurrent. During the first stage, patients present with viral symptoms, such as headaches, nausea, dizziness, and fever. During the acute uveitic stage, patients have decreased vision due to bilateral posterior uveitis and serous retinal detachments. During the convalescent stage, skin findings such as vitiligo, alopecia, and poliosis may be seen. Signs of hyper- or hypopigmentation in the eye may be present, including a “sunset-glow” fundus, depigmented chorioretinal lesions, called “Dalen Fuchs nodules,” and perilimbal vitiligo, known as Sugiura’s sign.(1,2,4) The chronic recurrent stage is characterized by episodes of panuveitis and development of iris nodules, posterior synechiae, glaucoma, cataracts, subretinal fibrosis, and chorioretinal atrophy.(1,4)
Standard treatment for VKH involves initiating oral prednisone (1-1.5 mg/kg/day) or intravenous methylprednisolone (1 g daily for 3 days) followed by a switch to oral prednisone, with a gradual taper over 3-6 months.(3) Relapses may occur if the patient is taken off corticosteroids too early. In cases where inflammation is inadequately controlled or recurrent, immunosuppressive therapy is usually initiated.
Some researchers have advocated using antimetabolites, cyclosporine, and biological agents as first-line and maintenance therapy for VKH, suggesting they may lead to better long-term visual outcomes.(1,3,4) Periocular steroid injections and intravitreal fluocinolone implants can be used as adjunctive therapy.(3,4)
With adequate treatment with corticosteroids, the prognosis for VKH is generally good. By following the treatment regimen with the necessary follow-ups, the majority of patients achieve a visual acuity of 20/40 or better and avoid complications, such as cataract, glaucoma, choroidal neovascularization, and subretinal fibrosis.(1)
2. Lavezzo MM, Sakata VM, Morita C, et al. Vogt-Koyanagi-Harada disease: review of a rare autoimmune disease targeting antigens melanocytes. Orphanet J Rare Dis 2016;11:29.
3. McCannel CA. Basic and Clinical Science Course (BCSC), Section 12: Retina and Vitreous. American Academy of Ophthalmology; 2015:223-33.
4. O’Keefe GAD, Rao NA, Vogt-Koyanagi-Harada disease. Surv Ophthalmol 2017;62:1-25.