27 year old woman with blurred central vision OS and headache
Digital Journal of Ophthalmology 1997
Volume 3, Number 18
May 15, 1997
Volume 3, Number 18
May 15, 1997
Yichieh Shiuey, MD | Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, MA
POHx: None
PMHx: Non-contributory
Meds: Ibuprofen
SHx: Non-contributory
FHx: Non-contributory
Pupils: 2+ left afferent pupillary defect
External exam: Normal
Motility: Full, slight increase in left periorbital pain with extreme right gaze
Slit lamp examination: Normal ou. Anterior chambers deep and quiet.
Intraocular pressure: 15 mm Hg OU
Visual field Examination: See Figures 1-2
Fundus examination: See Figure 3-4
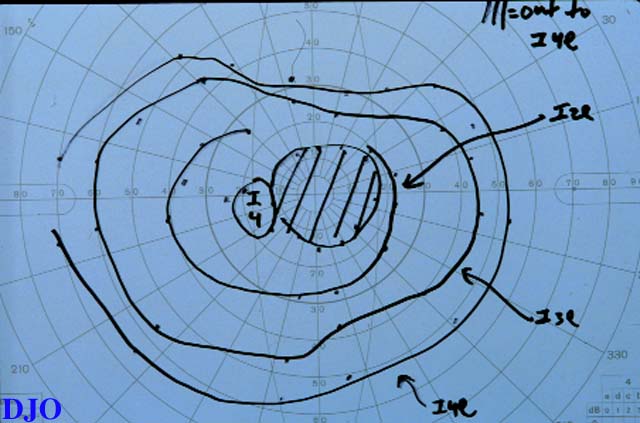
Figure 1
Figures 1-2. Goldmann visual fields are shown. There is a dense central scotoma OS. In addition, there was a paracentral visual field defect OD.
Figures 1-2. Goldmann visual fields are shown. There is a dense central scotoma OS. In addition, there was a paracentral visual field defect OD.
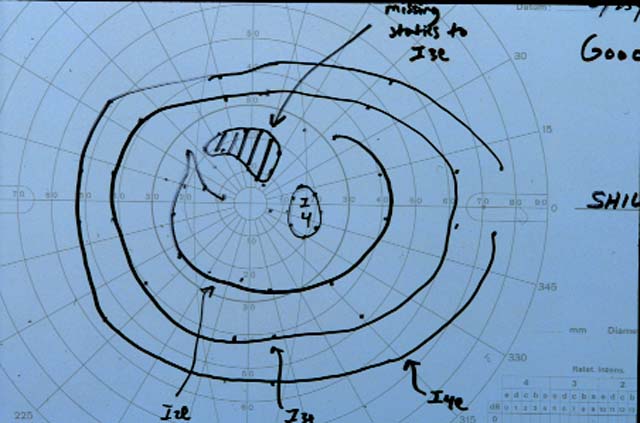
Figure 2
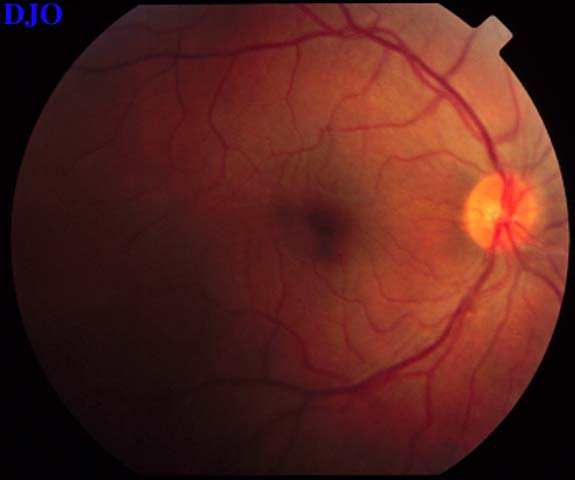
Figure 3
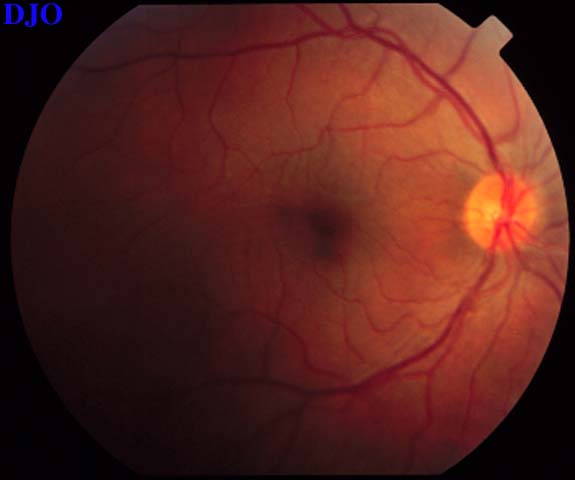
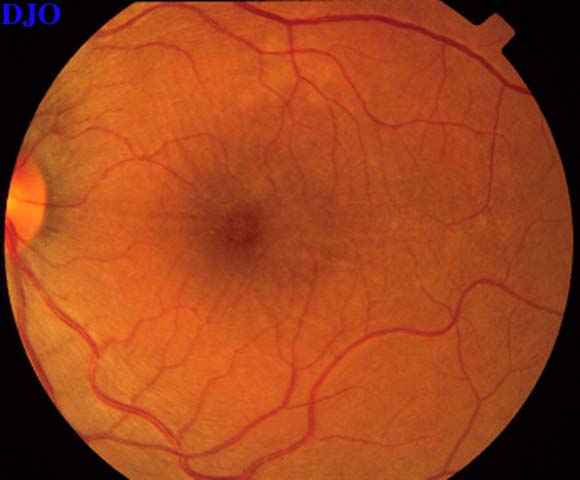
Figures 3-4. The right and left fundi are shown. The right fundus demonstrated fine striae in the macula, which were evident with slitlamp biomicroscopy. The left fundus showed an elevated serous retinal detachment of the macula and the peripapillary retina. The left disk appeared slightly hyperemic, but appeared otherwise normal.
Figures 3-4. The right and left fundi are shown. The right fundus demonstrated fine striae in the macula, which were evident with slitlamp biomicroscopy. The left fundus showed an elevated serous retinal detachment of the macula and the peripapillary retina. The left disk appeared slightly hyperemic, but appeared otherwise normal.
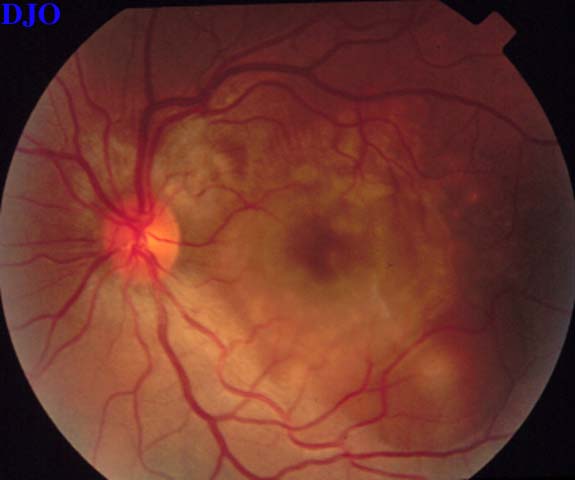
Figure 4
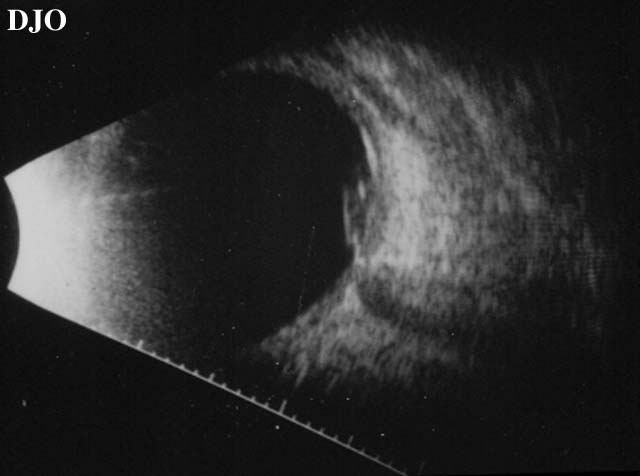
Figure 5
Demonstrates a serous retinal detachment at the macula seen on ultrasound.
Demonstrates a serous retinal detachment at the macula seen on ultrasound.
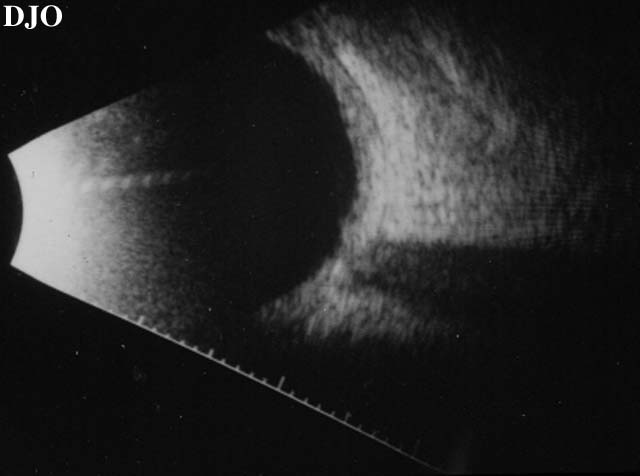
Figure 6
Shows thickening of the choroidal layer.
Shows thickening of the choroidal layer.
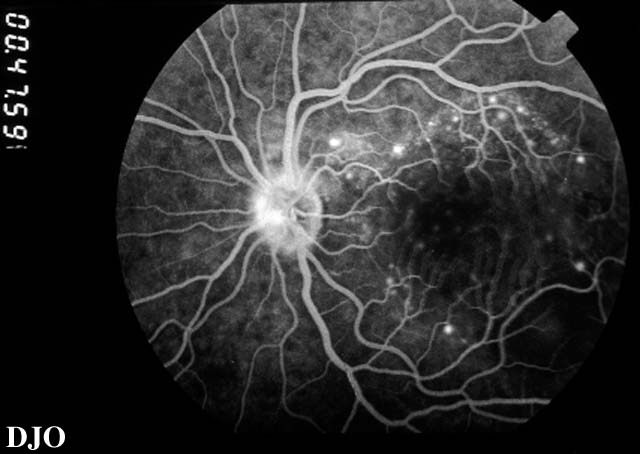
Figure 7
Early phase of the patient's fluorescein angiogram is shown. There are pinpoints of hyperfluorescence present.
Early phase of the patient's fluorescein angiogram is shown. There are pinpoints of hyperfluorescence present.
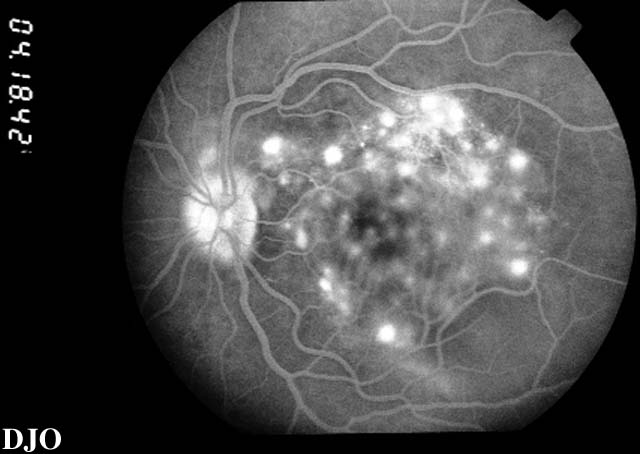
Figure 8
Late views of the angiogram are shown which demonstrate late leakage.
Late views of the angiogram are shown which demonstrate late leakage.
- Uveal effusion syndrome
- Tumor
- Harada's disease
Background:
Vogt in 1906 and Koyanagi in 1929 both described patients with bilateral anterior uveitis, vitiligo, poliosis, alopecia, and dysacusia. Harada in 1926 described patients with posterior uveitis, exudative retinal detachment, and cerebrospinal fluid pleocytosis. Over time it was recognized that there was significant overlap in the clinical characteristics of these patients and that these different clinical features were manifestations of the same disease. Hence, ophthalmologists began to call this constellation of clinical findings Vogt-Koyanagi-Harada syndrome or simply VKH. Some authors have also called this condition uveo-meningitic syndrome because of the frequent combination of uveitis and meningitis.
Epidemiology:
This condition has an equal gender distribution and usualy occurs between the ages of 20 to 50. VKH may occur in persons of any ethnic group, but is more common in Asians, Native Americans, and more darkly pigmented races. VKH is particularly common in Japan, where it represents 8% of all uveitis cases there. There is also an an association between VKH and certain HLA types including HLA-Bw54, HLA-DR4, and HLA-DqWa.
Clinical Features:
Although VKH is believed to be one single entitiy, it is clinically useful to differentiate between the Vogt-Koyanagi variant and the Harada's variant. The following TABLE points out some of the more important differences between the different possible presentations of VKH.
and psychosis
a "sunset glow"