29 year old man with visual symptoms and headaches
Digital Journal of Ophthalmology 1997
Volume 3, Number 14
March 4, 1997
Volume 3, Number 14
March 4, 1997
Yichieh Shiuey, MD | Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, MA
PMHx: Decreased hearing in both ears since childhood. He notes that he has had pre-mature graying since age 16. At age 16 he had a patch of gray hair on his forehead and gray hairs on his chest.
Meds: Non Contributory
SHx: Non contributory
FHx: He states there is a strong resemblance between him and his father. Otherwise no other family history.
Color: 8/8 Ishihara color plates identified correctly in each eye
Pupils: Round and equal in size, briskly reactive, No APD
External: See Figured 1-3
Slit lamp examination: See Figures 4-5
Fundus examination: Normal OU
Goldmann Visual Fields: Normal OU
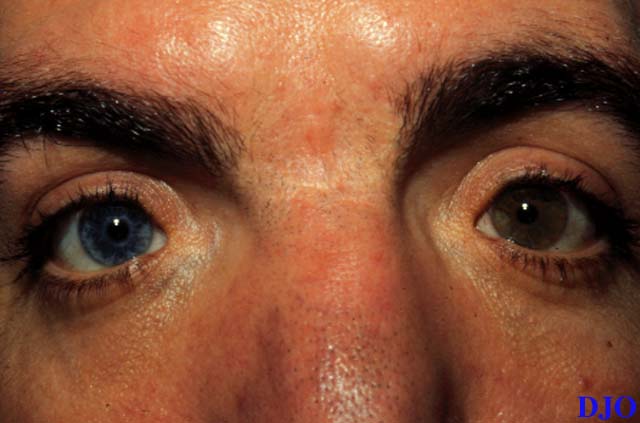
Figure 1
This patient had a broad nasal bridge. On close inspection the patient had an increased intercanthal distance of 47 mm with lateral displacement of the medial canthi. The lacrimal puncta were also laterally displaced, relative to the medial canthus. The patient had a normal interpupillary distance of 58 mm.
This patient had a broad nasal bridge. On close inspection the patient had an increased intercanthal distance of 47 mm with lateral displacement of the medial canthi. The lacrimal puncta were also laterally displaced, relative to the medial canthus. The patient had a normal interpupillary distance of 58 mm.
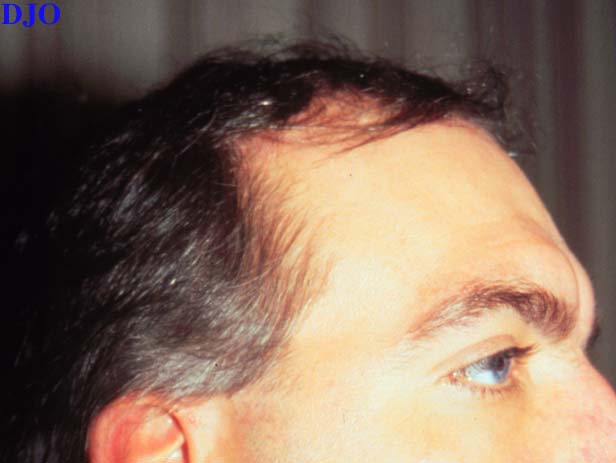
Figure 2
Figures 2-3. This 29 year old patient had greying of the hair near his right temple. On his left arm there was a patch of grey hair with a well circumscribed area of underlying skin hypopigmentation.
Figures 2-3. This 29 year old patient had greying of the hair near his right temple. On his left arm there was a patch of grey hair with a well circumscribed area of underlying skin hypopigmentation.
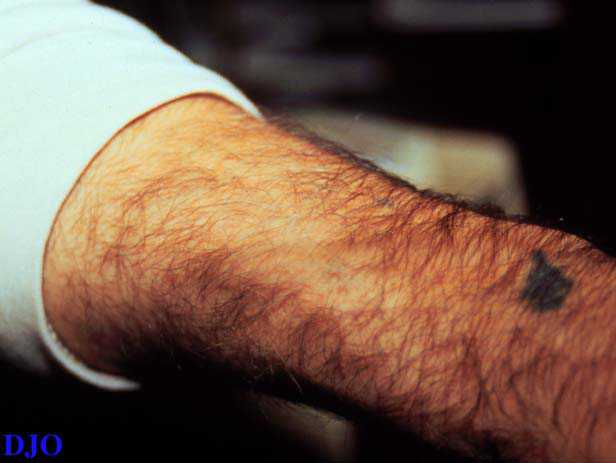
Figure 3
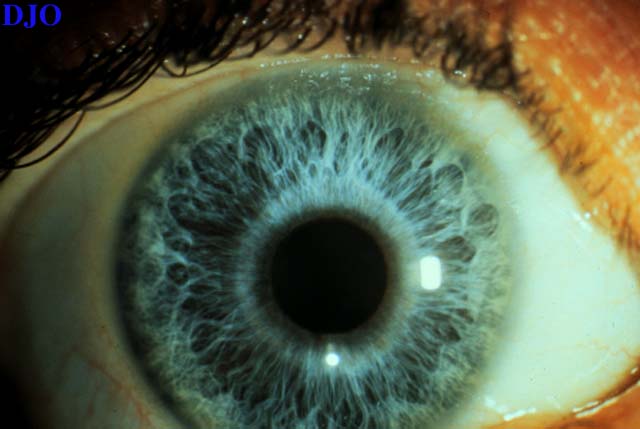
Figure 4
Figures 4-5. Iris heterochromia was present with a blue iris on the right and a green iris on the left.
Figures 4-5. Iris heterochromia was present with a blue iris on the right and a green iris on the left.
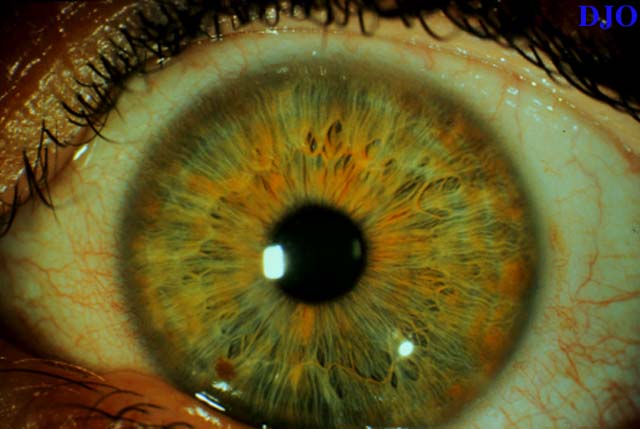
Figure 5
- Fuch's heterochromic cyclitis
- Retained intraocular foreign body
- Waardenburg's syndrome
- Ocular melanocytosis
- Diffuse iris nevus
- Iris melanoma
Our patient's symptoms and complete neuro-ophthalmic examination were felt to be consistent with migraine headaches. These symptoms were not believed to be associated with his incidental diagnosis of Waardenburg's syndrome. When asked if he was diagnosed with this condition before, he replied that he may have recalled a physician using that term when he was a teenager. The clinical entity of Waardenburg's syndrome was first described in 1951. In the original description this syndrome consisted of developmental abnormalities in the eyelids, eye brows, nose-root, associated with pigmentary defects of the irides and head hair and congenital deafness.
One of the cardinal findings of Waardenburg's syndrome is iris heterochromia. It is believed that this condition produces hypo-pigmentation of the affected iris as opposed to hyper-pigmentation. The evidence for this is the striking observation that patients with Waardenburg's syndrome of African descent have blue irides. Some patients may have hypo-chromic irides bilaterally without heterochromia. Hypopigmentation of the fundi may also be present.
Pigmentatry abnormalities of the hair and skin are another prominent feature of this syndrome. Classically, there is a white forelock of hair (which this patient had as a teenager), but premature graying of hair and patches of white body hair are also very common. Well circumscribed hypo-pigmentation of the skin may also be found and is termed leucism. Other hair abnormalities in Waardenburg's include fusion of the eyebrows (synophrys).
Telecanthus is the main eyelid abnormality caused by lateral displacement of the medial canthus. Lateral displacement of the punctum relative to the medial canthus occurs in Type I Waardenburg's syndrome. This is referred to as dystopia canthorum. Type II Waardenburg's syndrome is distinguished FROM type I by the absence of dystopia canthorum.
Unilateral or bilateral congenital deafness frequently occurs, but may be of varying severity. It has been observed that deafness is twice as common in type II Waardenburg's patients compared to type I.
Waardenburg's syndrome is an autosomal dominant condition. The gene mutation in Type I has been mapped to chromosome 2. Type II has been mapped to chromosome 3.
Most patients with this syndrome have normal vision and do not have nystagmus. Occasional associations of Waardenburg's syndrome include Hirschsprung's aganglionic megacolon and cleft lip or palate.
2) Baldwin CT, Hoth CF, Amos JA et al: An exonic mutation in the HuP2 paired domain gene causes Waardenburg's syndrome. Nature 355:637 (1993)