57 year old woman who complains "The lids are somewhat down"
Digital Journal of Ophthalmology 1997
Volume 3, Number 12
February 4, 1997
Volume 3, Number 12
February 4, 1997
Melanie Ryan-Graham, MD | Massachusetts Eye and Ear Infirmary, Harvard Medical School, Boston, MA
PMHx: Hypertension, rheumatoid arthritis
Meds: Xanax, Premarin, Provera, Methotrexate, Metoprolol, Calcium, Vitamin E
SHx: Non contributory
FHx: Paternal grandmother with ptosis
ROS:
- No diurnal variation of ptosis.
- No diplopia.
- No symptoms of Giant cell arteritis
- No generalized muscle weakness
- Dysphagia (solids > liquids) for many years, requiring Heimlich maneuver on several occasions
Color: Normal OU
Pupils: Light-near dissociation of the right pupil, no pupillary ruff OD, consistent with Adie's pupil. Left pupil normal.
Exophthalmometry: 12mm OU, base 98
Orbits: No resistance to retropulsion
Lids:
- Palpebral fissure: 5mm OU
- Marginal reflex distance: -1mm OU
- Levator function: 11mm OU
- Upper lid crease: 10.5 OU (normal)
- Orbicularis oculi: slight weakness OU
Motility: full, no diplopia. Good Bell's phenomenon
Neurologic: Corneal sensation intact, palatal speech, weak lower facial and neck muscles, transverse smile. No myotonia with handshake
Fundus: No pigmentary retinopathy
Visual Fields: See Figures 2a-2b
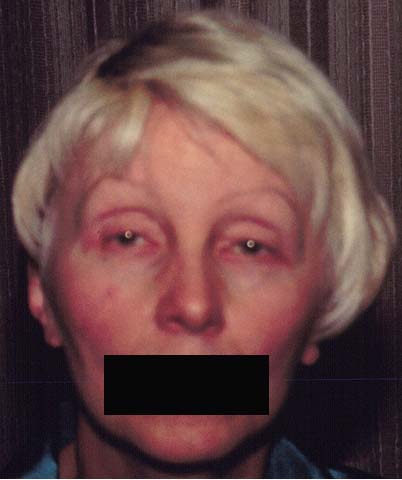
Figure 1
The clinical photo demonstrates the patient's ptosis
The clinical photo demonstrates the patient's ptosis
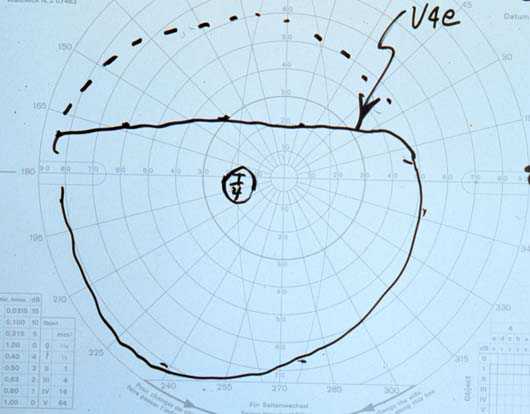
Figure 2a
Figures 2a-2b. The Goldmann visual field demonstrated a superior field defect OU which dissappeared with elevation of the upper lids.
Figures 2a-2b. The Goldmann visual field demonstrated a superior field defect OU which dissappeared with elevation of the upper lids.
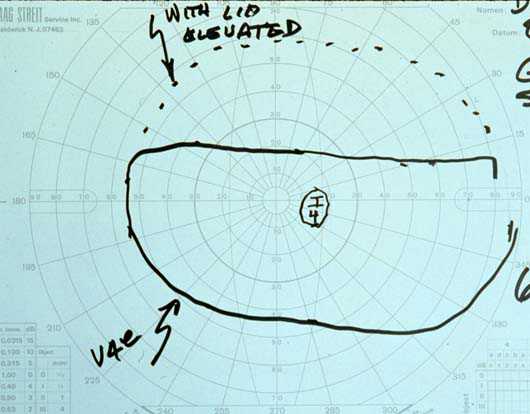
Figure 2b
Considering the history and examination findings, the differential was narrowed to myogenic ptosis and myasthenia gravis. Acquired myogenic ptosis includes myotonic dystrophy, chronic progressive external ophthalmoplegia, and oculopharyngeal dystrophy. Myotonic dystrophy is an autosomal dominant muscular dystropy with onset in the second decade of life, characterized by delayed relaxation of skeletal muscles after contraction. Symptoms include blepharospasm, which is followed by muscle atrophy, ptosis and weakness of orbicularis oculi. There are slow saccades and ophthalmoplegia often develops. The pupils are often miotic and sluggish to light and near. Characteristic Christmas tree cataracts (fine anterior and posterior subcapsular colored crystals) are found in virtually 100% of patients. There may be associated cardiac disease, dysphagia, constipation, incontinence, mental retardation, hyperostosis, scoliosis, testicular atrophy, and baldness.
Chronic progressive external ophthalmoplegia and the Kearns-Sayre syndrome are mitochondrial cytopathies characterized by adolescent onset bilateral asymmetric ptosis followed by progressive limitation of ocular motility, often with sparing of down-gaze. Examination findings often include slow saccades, nystagmus, and weak orbicularis oculi. In the Kearns-Sayre variant, there has been described pigmentary retinopathy, cardiac conduction abnormalities, ataxia, deafness, peripheral neuropathy, short stature, endocrine abnormalites including diabetes mellitus and hypogonadism, and elevated CSF protein. There are mitochondrial abnormalities on skeletal muscle biopsy, with ragged red fibers on Gomori's trichome stain and large irregular mitochondria with succinic dehydrogenase enzyme staining. Treatment includes coenzyme Q or ubidecarenone replacement to enhance mitochondrial respiration, which may improve cardiac function, ataxia, and exercise tolerance, but does not affect ophthalmplegia, ptosis, or retinopathy.
Oculopharyngeal dystrophy is an autosomal dominant muscular dystrophy that is most commonly found in French-Canadians with an onset of ptosis and dysphagia in the third or fourth decade. The dysphagia is due to weakness and lack of coordination of the muscles of the hypopharynx, resulting in an increased risk of aspiration pneumonia. There may also be some degree of ophthalmoplegia and weakness of the orbicularis, facial, and limb-girdle muscles.
Given the classic presentation of a French-Canadian woman with acquired bilateral ptosis, good extraocular motility, dysphagia with consequent aspiration pneumonia, mild lower facial muscle weakness, and a family history of ptosis, in the absence of pigmentary retinopathy, cataracts, and other systemic findings, the patient was diagnosed with oculopharyngeal dystrophy. She has been referred to an ENT specialist for dysphagia evaluation, and is being evaluated for ptosis repair.
Oculopharyngeal dystrophy was first described by Taylor in 1915 in a Boston family of French-Canadian descent. He described the syndrome as a progressive swallowing disorder with ptosis that eventually caused death by starvation. Victor named the syndrome "oculopharyngeal dystrophy" in 1962, although in Quebec it is known as "Barbeau's disease", named after Barbeau who published a report in1966 that traced the syndrome to a single couple that migrated in 1634 FROM France to Quebec and settled in the counties of LÕIslet and Montmagny along the St. Lawrence River. Over a hundred cases have been traced to this couple. Barbeau established the autosomal dominant inheritance of most cases, although there have been occasional autosomal recessive and sporadic cases reported. The syndrome is more common in persons of French-Canadian descent and has a high prevalence in Quebec, but has been reported in 20 other countries.
Generally, the syndrome presents with dysphagia that is followed by ptosis within a few years. Both liquids and solids may be difficult to swallow, and there is a tendency to aspiration. On examination, it is noted that the pharyngeal musculature is weak and uncoordinated, and that the cricopharyngeal muscle has abnormal reflex relaxation. There is indeed an associated weight loss with the progression of the dysphagia. Also, the altered pharyngeal and laryngeal musculature can cause a palatal voice.
The ptosis is generally bilateral and symmetric. In addition, there may be ophthalmoparesis, but generally of only mild degree. There may also be mild weakness in facial, neck flexor, and proximal limb muscles.
The etiology of the disease is unknown. Originally Taylor described it as the neurogenic disorder "progressive vagus-glossopharyngeal paralysis with ptosis". Victor in 1962 reported that there was evidence of a muscular dystrophy. Currently, most investigators suspect that it is a primary myopathy, although some still favor a neurogenic etiology. It is difficult to sort because EMG and pathologic studies of chronic denervation and myopathy can be similar. On pathologic examination, the skeletal muscle fibers are of decreased size and with fibrosis. Tubulofilamentous intranuclear inclusions of characteristic size and location described by TomŽ and colleagues can be detected by electron microscopy of skeletal muscle biopsies. These are pathognomonic of the disease although not present in all muslce fibers. Mitochondrial abnormalities have been noted, but are not specific and are not thought to be the primary disorder.
Management includes careful medical and family history, complete examination, and ENT referral. A Tensilon test should also be considered. Careful family history taking is helpful both for providing a basis for genetic counseling and for helping predict the patient's future course which is often similar to relatives. ENT evaluation is essential for the evaluation and management of dysphagia. The surgical treatment of the dysphagia consists of inferior constrictor myotomy. Before ptosis surgery is undertaken, careful documentation of good Bell's phenomenon, orbicularis function, and corneal sensation should be done. A study by Molgat and colleagues reports impressive success in levator resection for the surgical correction of ptosis due to oculopharyngeal muscular dystrophy.
In summary, oculopharyngeal muscular dystrophy is a disease that is commonly found among those of French-Canadian descent, generally has an autosomal dominant inheritance, is characterized by ptosis and dysphagia, has a high risk of aspiration pneumonia and starvation, and is often amenable to surgical correction of the dysphagia via inferior constrictor myotomy, and of the ptosis by levator resection.
2) Hardiman O, Halperin JJ, Farrell MA et al: Neuropathic Findings in Oculopharyngeal Muscular Dystrophy. Archives Neurology 50:481-488 (1993)
3) Molgat, Y, Rodrique D. Correction of blepharoptosis in oculoppharyngeal muscular dystrophy: Review of 91 cases. Canadian Journal of Ophthalmology 28:11-14 (1993)
4) Jordan DR, Addison DJ. Surgical results and pathological findings in the oculopharyngeal dystrophy syndrome. Canadian Journal of Ophthalmology 28:15-18 (1993)
5) DeWilde F, D'Haens M, Smet H, Martin JJ, Tassignon MJ. Surgical treatment of myogenic blepharoptosis. Bull. Soc. Belge Ophthalmol. 255: 139-146 (1994)
6) Fradat G, Pouliot D, Lavoie S, St-Pierre S. Inferior constrictor myotomy in oculopharyngeal muscular dystrophy: clinical and manometric evaluation. Journal of Otolaryngology 17:2, 68-73 (1988)