A 5-year-old girl with decreased vision in the left eye
Digital Journal of Ophthalmology 2015
Volume 21, Number 2
May 9, 2015
DOI: 10.5693/djo.03.2014.09.001
Volume 21, Number 2
May 9, 2015
DOI: 10.5693/djo.03.2014.09.001
Download PDF
Joel Yap, MBChB | Department of Ophthalmology, Greenlane Clinical Centre, Auckland District Health Board, New Zealand
Dianne Sharp, FRANZCO | Department of Ophthalmology, Greenlane Clinical Centre, Auckland District Health Board, New Zealand
Shuan Dai, FRANZCO | Department of Ophthalmology, Greenlane Clinical Centre, Auckland District Health Board, New Zealand
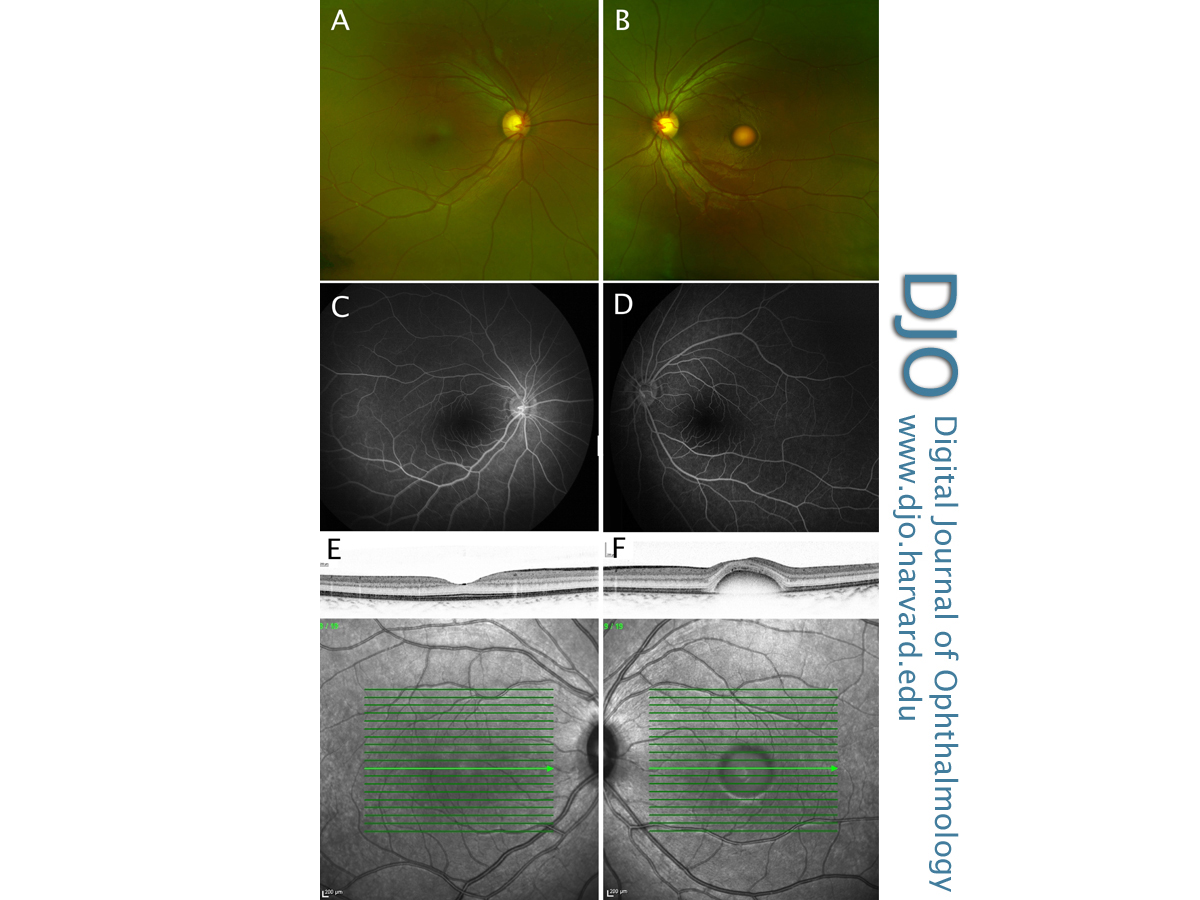
Figure 1
Color fundus photographs (A,B), late-stage fundus fluorescein angiogram (C,D) and infrared reflectance imaging with corresponding SD-OCT foveal cross section (E,F) of both the right and left eye, demonstrating asymmetric disease.
Color fundus photographs (A,B), late-stage fundus fluorescein angiogram (C,D) and infrared reflectance imaging with corresponding SD-OCT foveal cross section (E,F) of both the right and left eye, demonstrating asymmetric disease.
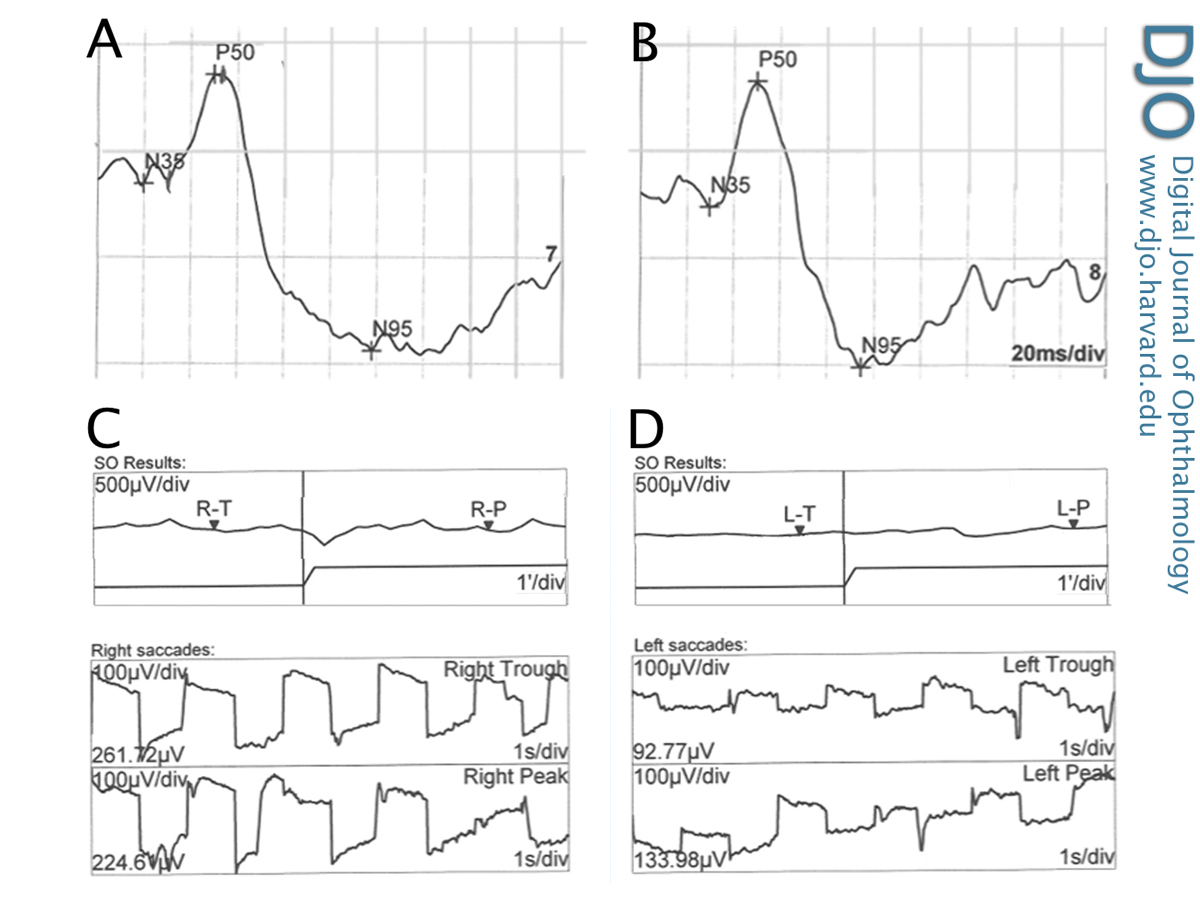
Figure 2
Electrophysiology encompasses several objective examination techniques that measure the function of the retina by measuring action potentials caused by particular patterns of light stimulation. Figure 2 shows a pattern ERG with well defined P50 and N95 components in both the right (A) and left (B) eyes, demonstrating symmetrical and normal macular function. Electrooculography measures the difference in electrical potential between the front and back of the eye, which is mostly a function of the retinal pigment epithelium. In this case, there are decreased light rise responses in both the right (C) and left (D) eyes. The Arden ratio (light to dark ratio) was reduced in both eyes.
Electrophysiology encompasses several objective examination techniques that measure the function of the retina by measuring action potentials caused by particular patterns of light stimulation. Figure 2 shows a pattern ERG with well defined P50 and N95 components in both the right (A) and left (B) eyes, demonstrating symmetrical and normal macular function. Electrooculography measures the difference in electrical potential between the front and back of the eye, which is mostly a function of the retinal pigment epithelium. In this case, there are decreased light rise responses in both the right (C) and left (D) eyes. The Arden ratio (light to dark ratio) was reduced in both eyes.
Subash et al published a retrospective case series of 6 patients with unilateral vitelliform maculopathy.(2) The patients ranged in age from 30 to 68 years. In addition to performing ocular examination and obtaining ocular investigations, they acquired blood samples for DNA extraction and mutation screening of the BEST1 and PRPH2 genes. Molecular screening of both genes revealed no mutations in any patient. Interestingly, the electrooculography light-rise and full-field ERG were within normal limits for all of their patients. Our patient differed from the patients in their case series because she was much younger and had an abnormal electrooculogram.
Cascavilla et al reported a similar case, involving an 8-year-old girl.(3) She also presented with mildly reduced vision in the left eye and was discovered to have a round, yellow macular lesion in that eye. However, SD-OCT and fundus color photographs of the lesion showed more heterogeneity than in the present case. Additionally, their patient developed full-field ERG delayed rod and cone responses. On gene analysis, the patient was found to have a homozygous mutation in the BEST1 gene, suggesting an autosomal recessive inheritance pattern.
The BEST1 gene is virtually the only gene involved in BVMD. It encodes a 68-kDA protein bestrophin-1, which is thought to function as a chloride channel localized to the basolateral plasma membrane of the retinal pigment epithelium (RPE), which is critical for the phagocytosis of shed outer segments and is the site for the regeneration of the chromophore 11-cis-retinal. Over 200 different BEST1 mutations have been described in families affected by BVMD.(1,4)
Previous studies have suggested that patients with an autosomal recessive pattern of inheritance in mutations of the BEST1 gene have a phenotype that is different from those with autosomal dominant disease. The most commonly reported distinguishing features are extrafoveal and extramacular subretinal deposits. Kinnick et al reported that patients with compound heterozygous mutations also have phenotypes that differ from classic dominant BVMD, in that macular deposits tended to be inferior to the fovea, and some degree of subretinal fibrosis was present.(1) About half of their studied patients had small, round, yellow or white spots that were most prominent along the temporal vascular arcades.
Zhang et al performed a post-mortem histological analysis on the macula of a patient with BVMD and found that the clinical finding of an elevated submacular yellow lesion corresponded histologically to RPE hyperplasia, accumulation of lipofuscin in the RPE, and deposition of granular material in the photoreceptors and macrophages.(5) Kay and colleagues, utilizing SD-OCT, suggested that the vitelliform material is located in the subretinal space and concluded that the disease is associated with diffuse photoreceptor outer segment abnormalities overlying a structurally normal RPE.(6)
Acknowledgments
The authors thank the clinical staff at Greenlane Clinical Centre, Auckland, for the images used to document this case.
2. Subash M, Rotsos T, Wright GA, et al. Unilateral vitelliform maculopathy: a comprehensive phenotype study with molecular screening of BEST1 and PRPH2. Br J Ophthalmol 2012;96:719-22.
3. Cascavilla ML, Quergues G, Stenirri S, Battaglia Parodi M, Quergues L, Bandello F. Unilateral vitelliform phenotype in autosomal recessive bestophinopathy. Ophthalmic Res 2012;48:146-50.
4. Burgess R, Millar I, Leroy B, Urquhart J, Fearon I, Baere E, Brown P, Robson A, Wright G, Kestelyn P, Holder G, Webster A, Mandon F and Black G. Biallelic mutation BEST1 causes a distinct retinopathy in humans. Am J Hum Genet 2008; 82:19-31.
5. Zhang Q, Small K and Grossniklaus H. Clinicopathological findings in Best vitelliform macular dystrophy. Graefes Arch Clin Exp Ophthalmol 2011;249:745-51.
6. Kay C, Abramoff M, Mullins R, Kinnick T, Lee K, Eyestone M, Chung M, Sohn E and Stone E. Three-dimensional distribution of the vitelliform lesion, photoreceptors, and retinal pigment epithelium in the macula of patients with best vitelliform macular dystrophy. Arch Ophthalmol 2012;130:357-64.