A 48-year-old man presents with bilateral corneal deposits
Digital Journal of Ophthalmology 2009
Volume 15, Number 4
November 27, 2009
Volume 15, Number 4
November 27, 2009
Download PDF
Shaminder S. Bhullar, MD | Krieger Eye Institute, Sinai Hospital
Gerami D. Seitzman, MD | Krieger Eye Institute, Sinai Hospital
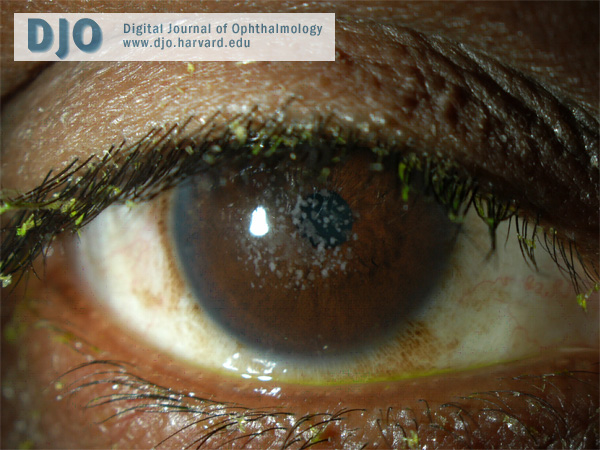
Figure 1
External photo of the right eye.
External photo of the right eye.
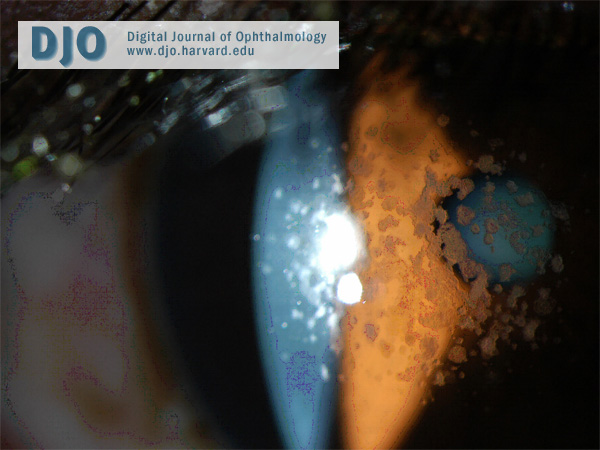
Figure 2
In type 2 granular corneal dystrophy, the “bread crumb” lesions coalesce into fewer larger ring-shaped granular deposits with sharply defined intervening clear zones.
In type 2 granular corneal dystrophy, the “bread crumb” lesions coalesce into fewer larger ring-shaped granular deposits with sharply defined intervening clear zones.

Figure 3
Slit-lamp photograph demonstrating anterior stromal location of the deposits.
Slit-lamp photograph demonstrating anterior stromal location of the deposits.
Our case is unusual in that our patient presented with no family history of any corneal dystrophy. Granular corneal dystrophy classically has an autosomal dominant inheritance pattern with strong penetrance. This dystrophy has been linked to a locus on region 5q22-32 on chromosome 5, as part of the BIGH3 complex, coding for keratoepithelin.(2) That our patient did not have a family history of this autosomal dominant disease may be explained by recent reports of de novo mutations in the BIGH3 gene, which have led to the development of granular corneal dystrophy.(3) Alternatively, this patient’s family members may harbor a gene mutation, and due to phenotypic non-penetrance associated with type 2 granular corneal dystrophy, may not manifest the disease.(4)
Granular corneal dystrophy is an uncommon disease without any gender predilection. It is more commonly present in Caucasian individuals. Our patient is African American, and it is less commonly seen in this population.(5) Although lesions may present earlier in life, vision is typically unaffected until the third or fourth decade. Some patients may complain of mild photophobia from light scatter due to the opacities. Ocular surface pain from recurrent corneal erosions occurs more commonly in the subset of patients in whom Bowman’s layer is involved.
There are three clinical forms of granular corneal dystrophy. Type 1 is the most frequent. It occurs early in life with crumb-like opacities that may slowly progress into disc-like lesions. The lesions in this variant may extend anteriorly to involve Bowman’s layer. Type 2 patients typically present in their second decade, with fewer larger ring-shaped granular deposits in the anterior stroma with sharply defined intervening clear zones. Erosions are infrequent in this variant and vision is unaffected because of the intervening clear areas. The opacities may extend posteriorly as the patient ages. The third type of granular corneal dystrophy presents in infancy with superficial lesions, confined to Bowman’s layer, resembling Reis-Bücklers corneal dystrophy. Our patient has type 2 granular corneal dystrophy. His slit-lamp examination discloses large, sharp, ring-shaped granular deposits with intervening clear zones. He has retained good visual acuity and has fortunately not developed recurrent erosions.
In the early stages of granular dystrophy no treatment is needed. In patients who develop recurrent erosions, symptomatic treatment with lubricating ophthalmic ointment, bandage contact lenses, superficial keratectomy or phototherapeutic keratectomy may be used. Visual acuity is typically affected when the intervening clear zones begin to take on a ground glass appearance. When visual acuity is affected, penetrating keratoplasty or deep anterior lamellar keratoplasty can be considered.(6) Recurrences of the dystrophy may occur and typically appear as diffuse superficial subepithelial lesions in the periphery.
2. Korvatska E, Munier FL, Djemaï A., et al. Mutation hot spots in 5q31-linked corneal dystrophies. Am J Hum Genet 1998;62(2):320-4.
3. Hilton EN, Black GC, Manson FD, et al. De novo mutation in the BIGH3/TGFB1 gene causing granular corneal dystrophy. Br J Ophthalmol 2007;91(8):1083-4.
4. Kim JW, Kim HM, Song JS. Phenotypic non-penetrance in granular corneal dystrophy type II. Graefes Arch Clin Exp Ophthalmol 2008;246(11):1629-31.
5. Meallet MA, Affeldt JA, McFarland TJ, et al. An unusual clinical phenotype of Avellino corneal dystrophy associated with an Arg124His beta iG-H3 mutation in an African-American woman. Am J Ophthalmol 2004;137(4);765-7.
6. Salouti R, Hosseini H, Eghtedari M, Khalili MR. Deep anterior lamellar keratoplasty with Melles technique for granular corneal dystrophy. Cornea 2009;28(2):140-3.