A 19-month-old girl with nystagmus, paradoxical pupillary response and low vision
Digital Journal of Ophthalmology 2008
Volume 14, Number 6
February 25, 2008
Volume 14, Number 6
February 25, 2008
The pupils were round and equal and constricted in response to light. No afferent pupillary defect was seen. Additionally, using an infrared video system, paradoxical pupillary constriction to darkness was demonstrated. (see Video)
The cycloplegic retinoscopy was +5.50 sphere in both eyes. She was orthotropic on muscle balance testing. She had full ductions and versions. There was a small amplitude, high frequency, horizontal nystagmus. The direction of nystagmus did not change in different directions of gaze. There was no anomalous head posture and the patient did not appear to have a null point in which the nystagmus dampened.
The anterior segment examination was normal. There were no iris transillumination defects. The intraocular pressures were normal at 8 mmHg in both eyes. On visual field testing, she looked for the hand light in all quadrants with both eyes. On indirect ophthalmoscopy, the media were clear; the optic nerve heads were pink with sharp margins and were normal in diameter. In the maculae, foveal reflexes were identifiable. The retinal vasculature appeared to be of normal caliber and distribution and no abnormalities were seen in the extramacular fundi. (see Figures 1 & 2)
Video
Infrared video system demonstrating the paradoxical pupillary constriction to darkness.
Infrared video system demonstrating the paradoxical pupillary constriction to darkness.
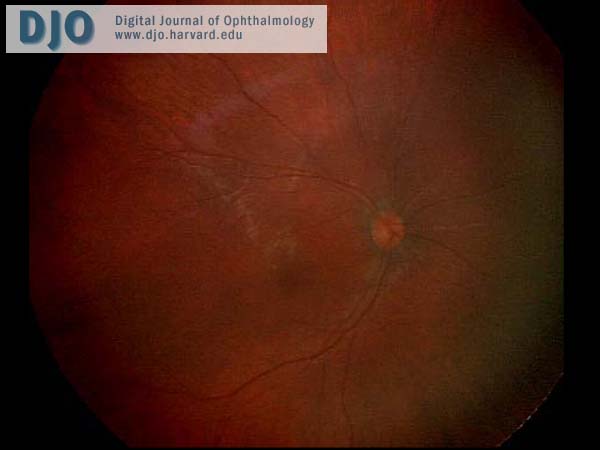
Figure 1
Ret Cam photograph of the right fundus.
Ret Cam photograph of the right fundus.
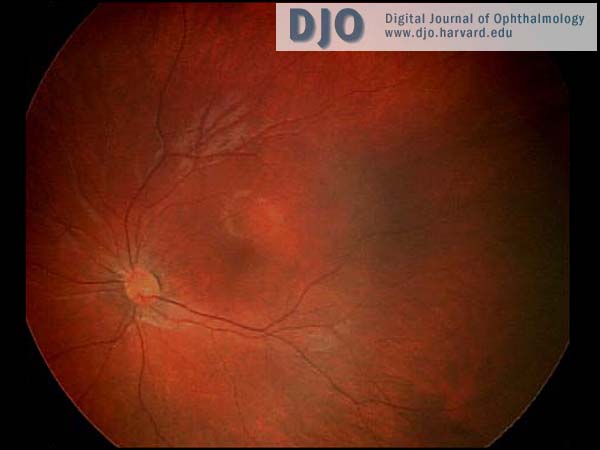
Figure 2
Ret Cam photograph of the left fundus.
Ret Cam photograph of the left fundus.
An electroretinogram (ERG) was performed and showed that the cone and cone-driven responses were absent or markedly attenuated. The rod responses were present but mildly attenuated. (See Figure 3)
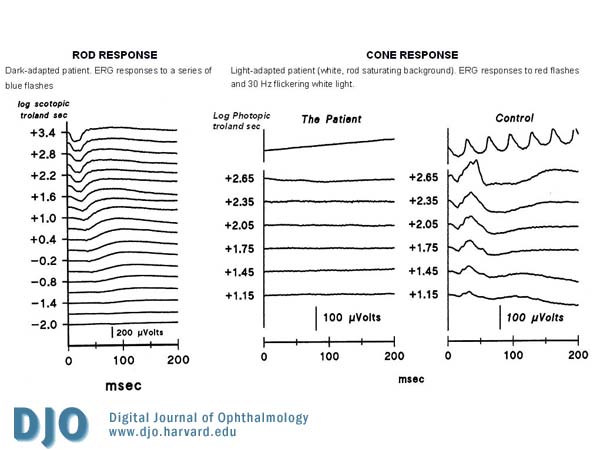
Figure 3
Electroretinogram (ERG) showing the absence of cone and cone-driven responses with decreased rod responses.
Electroretinogram (ERG) showing the absence of cone and cone-driven responses with decreased rod responses.
Achromatopsia ("a-", lack of; "-chroma-", color; "-opsia", vision), also called rod monochromacy is a stationary autosomal recessive condition that affects 8,500 persons in the US (1 in 33,000). It is characterized by reduced visual acuity, nystagmus, deficits in color discrimination, normal fundus appearance and paradoxical pupillary constriction to dark.(1,2) Hyperopia is common in individuals with achromatopsia, although a broad distribution of refractive errors has been reported.(3) Patients with achromatopsia are usually photophobic (light sensitive). Our patient does not appear to be photophobic, but she is not yet ambulatory. Some children with achromatopsia do not have photophobia until walking on their own outdoors.
Achromatopsia is caused by a channelopathy of the photoreceptors.(4) The most frequent molecular cause are mutations in the cGMP-gated cation channel genes CNGA3 (Chrom 2) and CNGB3 (Chrom 8) (5,6) and less commonly, a mutation in the transducin protein GNAT2.(7) As a result, cone and cone-driven ERG responses to full-field stimuli are markedly attenuated or non-detectable, but rod and rod-driven responses are normal or mildly decreased.(8,9)
In our patient although achromatopsia is the most likely diagnosis, a progressive retinal disorder cannot be ruled out completely. For this reason regular ophthalmic follow-up is recommended.
Treatment and Recommendations:
Patients should be prescribed glasses if there is a significant refractive error. Our patient was given +5.50 spherical refraction in both eyes. In our patient no additional treatment was offered at this point since she was not light sensitive and she is not ambulatory yet. She needs a follow up examination and the vision should be monitored with special tests that include measurement of the dark-adapted visual threshold and mapping of the visual fields. Other recommendations for patients with Achromatopsia include: Red/dark central contact lenses; sun filters to be worn outdoors to decrease light sensitivity and low vision aids including the use of large print materials, sitting in the classroom away from the windows and closing blinds to decrease glare on the board.
Acknowledgements
I would like to thank Anne Fulton, M.D. for her assistance in the preparation of this manuscript.
2. Ben Simon GJ, Abraham FA, Melamed S. Pingelapese achromatopsia: correlation between paradoxical pupillary response and clinical features. Br J Ophthalmol. 2004;88(2):223-5.
3. Haegerstrom-Portnoy G, Schneck ME, Verdon WA, Hewlett SE. Clinical vision characteristics of the congenital achromatopsias. I. Visual acuity, refractive error, and binocular status. Optom Vis Sci. 1996;73(7):446-56.
4. Kohl S, Marx T, Giddings I, et al. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet. 1998;19(3):257-9.
5. Wissinger B, Gamer D, Jagle H, et al. CNGA3 mutations in hereditary cone photoreceptor disorders. Am J Hum Genet. 2001;69(4):722-37.
6. Kohl S, Varsanyi B, Antunes GA, et al. CNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur J Hum Genet. 2005;13(3):302-8.
7. Aligianis IA, Forshew T, Johnson S, et al. Mapping of a novel locus for achromatopsia (ACHM4) to 1p and identification of a germline mutation in the alpha subunit of cone transducin (GNAT2). J Med Genet. 2002;39(9):656-60.
8. Andreasson S, Tornqvist K. Electroretinograms in patients with achromatopsia. Acta Ophthalmol (Copenh). 1991;69(6):711-6.
9. Michaelides M, Hunt DM, Moore AT. The cone dysfunction syndromes. Br J Ophthalmol. 2004;88(2):291-7.